Electrical energy storage for the grid: A battery of choices
1
2011
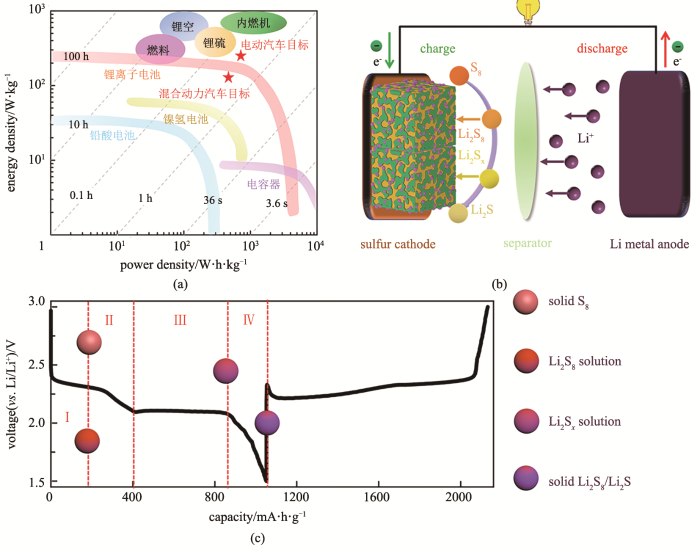
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Bridging the academic and industrial metrics for next-generation practical batteries
0
2019
Towards greener and more sustainable batteries for electrical energy storage
1
2015
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Freestanding Mo2C-decorating N-doped carbon nanofibers as 3D current collector for ultra-stable Li-S batteries
1
2019
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Novel sulfur host composed of cobalt and porous graphitic carbon derived from mofs for the high-performance Li-S battery
2
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
3D porous carbon sheets with multidirectional ion pathways for fast and durable lithium-sulfur batteries
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Electrolyte regulation towards stable lithium metal anode in lithium-sulfur batteries with sulfurized polyacrylonitrile cathode
1
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
A high-energy sulfur cathode in carbonate electrolyte by eliminating polysulfides via solid-phase lithium-sulfur transformation
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Chemisorption of polysulfides through redox reactions with organic molecules for lithium-sulfur batteries
0
2018
Facilitation of sulfur evolution reaction by pyridinic nitrogen doped carbon nanoflakes for highly-stable lithium-sulfur batteries
0
2018
锂硫电池硫正极技术研究进展
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
锂硫电池硫正极技术研究进展
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Popcorn inspired porous macrocellular carbon: Rapid puffing fabrication from rice and its applications in lithium-sulfur batteries
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Catalytic effects in lithium-sulfur batteries: Promoted sulfur transformation and reduced shuttle effect
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Superhierarchical cobalt-embedded nitrogen-doped porous carbon nanosheets as two-in-one hosts for high-performance lithium-sulfur batteries
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
柔性锂硫电池的材料设计与实现
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
柔性锂硫电池的材料设计与实现
1
2018
... 为了应对可充电式电子设备的迅猛发展,构筑高能量密度、低制作成本和长使用寿命的电化学储能器件是科研工作者们努力的方向[1-3].作为一种以单质硫为正极,锂金属为负极的二次电池,锂硫电池凭借其超高理论能量密度(2600 W·h/kg),相比于商业锂离子电池(140~260 W·h/kg)有着较大的优势[图1(a)] [4-5].除此之外,硫元素本身具有储量丰富、价格低廉、环境友好、兼具易取得性和可持续性等优点[6],被认为是极具发展潜力的储锂正极材料[7].但是,传统锂硫电池存在的一些缺点导致其难以实现规模化生产:①硫单质(5×10-30 S/cm)和最终反应产物Li2S(10-30 S/cm)的本征绝缘性使得其在储锂过程中具有缓慢的反应动力学,限制了活性硫的利用率;②中间产物长链多硫化物易溶于醚类电解液中,从而迁移到负极,引起“穿梭效应”,导致活性硫的流失和锂金属负极的腐蚀;③锂化过程中活性位点的体积膨胀导致结构崩塌[8-11].为了解决上述问题,近年来的研究主要集中于以下3个方面:①掺入具有高电导率的金属元素(Fe、Co、Ni、Se、Te等)加快电子传输和离子扩散速率;②分别采用物理化学策略,引入微孔碳/高极性的活性位点,通过“碳孔藏硫”的物理限域和极性位点的化学吸附来限制多硫化物的“穿梭效应”;③设计特殊的形貌结构来缓解锂化过程中活性位点的膨胀[12-13].虽然上述策略在一定程度上延缓了C/S体系循环寿命,但是目前锂硫正极的循环稳定性仍达不到市场需求[14].基于此,本文综述了一种聚合物衍生锂硫电极.将聚合物在高温下与单质链状硫环合作为锂硫电池正极材料,能有效改善传统锂硫体系循环稳定性差等问题,同时为聚合物衍生有机硫正极作为下一代可充电器件的研究前景进行了展望[15]. ...
Lithium sulfur battery oxidation/reduction mechanisms of polysulfides in THF solutions
1
1988
... 锂硫电池的早期研究可追溯至1988年,Peled等[16]将多硫化锂溶于四氢呋喃,并滴涂在玻碳电极上,在-2.0~1.3 V的电势窗,2~200 mV/s的扫描速率下进行循环伏安测试:检测到一个氧化峰和三个还原峰,证明多硫化锂的氧化还原反应是可逆的.从此以后,锂硫电池凭借其超高能量密度引起了越来越多科研工作者的关注[17].锂硫电池是实现单质硫和Li2S/Li2S2之间的相互转化的能量存储器件,但是与锂离子电池的“摇椅式”嵌入/脱嵌储能机理却不尽相同[18].在首次放电过程中,单质硫得到电子,形成易溶于醚类电解液中的链状多硫化锂,随着电解液扩散经隔膜到达负极.在室温下,典型的锂硫电池放电曲线由高电位和低电位两个平台组成,其放电过程可分为四个阶段[图1(b)~(c)][19].第一阶段:在高电位平台(约2.3 V),对应着硫单质(S8)开环形成长链多硫化物(Li2S8)的固/液双相还原过程;第二阶段:长链多硫化物(Li2S8)向短链多硫化物(Li2Sx,4≤x≤8)转变的液/液单相还原过程;第三阶段:在低电位平台(约2.0 V),溶解的短链多硫化物(Li2Sx)向难溶的Li2S2/Li2S转化的液/固双相还原过程;第四阶段:不溶的Li2S2向Li2S转变的固/固单相还原过程.其中,第一和第二阶段由于自放电对整个体系的容量贡献较少,第三阶段对储锂过程发挥主要贡献作用,具体的放电深度和容量贡献如表1所示. ...
A bifunctional perovskite promoter for polysulfide regulation toward stable lithium-sulfur batteries
1
2018
... 锂硫电池的早期研究可追溯至1988年,Peled等[16]将多硫化锂溶于四氢呋喃,并滴涂在玻碳电极上,在-2.0~1.3 V的电势窗,2~200 mV/s的扫描速率下进行循环伏安测试:检测到一个氧化峰和三个还原峰,证明多硫化锂的氧化还原反应是可逆的.从此以后,锂硫电池凭借其超高能量密度引起了越来越多科研工作者的关注[17].锂硫电池是实现单质硫和Li2S/Li2S2之间的相互转化的能量存储器件,但是与锂离子电池的“摇椅式”嵌入/脱嵌储能机理却不尽相同[18].在首次放电过程中,单质硫得到电子,形成易溶于醚类电解液中的链状多硫化锂,随着电解液扩散经隔膜到达负极.在室温下,典型的锂硫电池放电曲线由高电位和低电位两个平台组成,其放电过程可分为四个阶段[图1(b)~(c)][19].第一阶段:在高电位平台(约2.3 V),对应着硫单质(S8)开环形成长链多硫化物(Li2S8)的固/液双相还原过程;第二阶段:长链多硫化物(Li2S8)向短链多硫化物(Li2Sx,4≤x≤8)转变的液/液单相还原过程;第三阶段:在低电位平台(约2.0 V),溶解的短链多硫化物(Li2Sx)向难溶的Li2S2/Li2S转化的液/固双相还原过程;第四阶段:不溶的Li2S2向Li2S转变的固/固单相还原过程.其中,第一和第二阶段由于自放电对整个体系的容量贡献较少,第三阶段对储锂过程发挥主要贡献作用,具体的放电深度和容量贡献如表1所示. ...
Spherical macroporous carbon nanotube particles with ultrahigh sulfur loading for lithium-sulfur battery cathodes
1
2018
... 锂硫电池的早期研究可追溯至1988年,Peled等[16]将多硫化锂溶于四氢呋喃,并滴涂在玻碳电极上,在-2.0~1.3 V的电势窗,2~200 mV/s的扫描速率下进行循环伏安测试:检测到一个氧化峰和三个还原峰,证明多硫化锂的氧化还原反应是可逆的.从此以后,锂硫电池凭借其超高能量密度引起了越来越多科研工作者的关注[17].锂硫电池是实现单质硫和Li2S/Li2S2之间的相互转化的能量存储器件,但是与锂离子电池的“摇椅式”嵌入/脱嵌储能机理却不尽相同[18].在首次放电过程中,单质硫得到电子,形成易溶于醚类电解液中的链状多硫化锂,随着电解液扩散经隔膜到达负极.在室温下,典型的锂硫电池放电曲线由高电位和低电位两个平台组成,其放电过程可分为四个阶段[图1(b)~(c)][19].第一阶段:在高电位平台(约2.3 V),对应着硫单质(S8)开环形成长链多硫化物(Li2S8)的固/液双相还原过程;第二阶段:长链多硫化物(Li2S8)向短链多硫化物(Li2Sx,4≤x≤8)转变的液/液单相还原过程;第三阶段:在低电位平台(约2.0 V),溶解的短链多硫化物(Li2Sx)向难溶的Li2S2/Li2S转化的液/固双相还原过程;第四阶段:不溶的Li2S2向Li2S转变的固/固单相还原过程.其中,第一和第二阶段由于自放电对整个体系的容量贡献较少,第三阶段对储锂过程发挥主要贡献作用,具体的放电深度和容量贡献如表1所示. ...
Tuning the electrolyte network structure to invoke quasi-solid state sulfur conversion and suppress lithium dendrite formation in Li-S batteries
1
2018
... 锂硫电池的早期研究可追溯至1988年,Peled等[16]将多硫化锂溶于四氢呋喃,并滴涂在玻碳电极上,在-2.0~1.3 V的电势窗,2~200 mV/s的扫描速率下进行循环伏安测试:检测到一个氧化峰和三个还原峰,证明多硫化锂的氧化还原反应是可逆的.从此以后,锂硫电池凭借其超高能量密度引起了越来越多科研工作者的关注[17].锂硫电池是实现单质硫和Li2S/Li2S2之间的相互转化的能量存储器件,但是与锂离子电池的“摇椅式”嵌入/脱嵌储能机理却不尽相同[18].在首次放电过程中,单质硫得到电子,形成易溶于醚类电解液中的链状多硫化锂,随着电解液扩散经隔膜到达负极.在室温下,典型的锂硫电池放电曲线由高电位和低电位两个平台组成,其放电过程可分为四个阶段[图1(b)~(c)][19].第一阶段:在高电位平台(约2.3 V),对应着硫单质(S8)开环形成长链多硫化物(Li2S8)的固/液双相还原过程;第二阶段:长链多硫化物(Li2S8)向短链多硫化物(Li2Sx,4≤x≤8)转变的液/液单相还原过程;第三阶段:在低电位平台(约2.0 V),溶解的短链多硫化物(Li2Sx)向难溶的Li2S2/Li2S转化的液/固双相还原过程;第四阶段:不溶的Li2S2向Li2S转变的固/固单相还原过程.其中,第一和第二阶段由于自放电对整个体系的容量贡献较少,第三阶段对储锂过程发挥主要贡献作用,具体的放电深度和容量贡献如表1所示. ...
锂硫电池安全性问题现状及未来发展态势
1
2018
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
锂硫电池安全性问题现状及未来发展态势
1
2018
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
高体积能量密度锂硫电池的构建材料和电极
1
2017
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
高体积能量密度锂硫电池的构建材料和电极
1
2017
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
A polysulfide reduction accelerator—NiS2-modified sulfurized polyacrylonitrile as a high performance cathode material for lithium-sulfur batteries
1
2017
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
Stringed “tube on cube” nanohybrids as compact cathode matrix for high-loading and lean-electrolyte lithium-sulfur batteries
1
2018
... 图1(c)为典型的锂硫电池首次充放电曲线,出现了两处明显的平台转折点(约2.28 V和约2.05 V)[20].第一处对应着S—S键的断裂和聚硫离子增加,此时电解液的黏度迅速提升,产生了严重的动力学极化作用[21];第二处对应着不溶性的Li2S2/Li2S向易溶性的多硫化物的相转变,碳表面的Li2S2/Li2S固态层被氧化成多硫化物并溶于电解液中,导致极化程度下降[22-23]. ...
锂硫电池的穿梭效应与抑制
1
2017
... 虽然锂硫电池具有超高的理论能量密度,但在实际应用中却因固有的电导率低、多硫化物易穿梭等不可忽视的问题,导致其可实现的容量并不高.另外在循环过程中的正极材料储锂能力快速下降,循环寿命短,限制了锂硫电池实现商业化[24].因此,亟待开发出一种能有效提升活性硫的利用率,真正解决传统锂硫电池体系稳定性差,循环寿命短的新型锂硫电池体系[25-26].聚合物衍生锂硫电池体系相对于传统锂硫体系,以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是简单的物理负载(多孔碳载硫),故有着极大的应用前景[27]. ...
锂硫电池的穿梭效应与抑制
1
2017
... 虽然锂硫电池具有超高的理论能量密度,但在实际应用中却因固有的电导率低、多硫化物易穿梭等不可忽视的问题,导致其可实现的容量并不高.另外在循环过程中的正极材料储锂能力快速下降,循环寿命短,限制了锂硫电池实现商业化[24].因此,亟待开发出一种能有效提升活性硫的利用率,真正解决传统锂硫电池体系稳定性差,循环寿命短的新型锂硫电池体系[25-26].聚合物衍生锂硫电池体系相对于传统锂硫体系,以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是简单的物理负载(多孔碳载硫),故有着极大的应用前景[27]. ...
Identifying the components of the solid-electrolyte interphase in Li-ion batteries
1
2019
... 虽然锂硫电池具有超高的理论能量密度,但在实际应用中却因固有的电导率低、多硫化物易穿梭等不可忽视的问题,导致其可实现的容量并不高.另外在循环过程中的正极材料储锂能力快速下降,循环寿命短,限制了锂硫电池实现商业化[24].因此,亟待开发出一种能有效提升活性硫的利用率,真正解决传统锂硫电池体系稳定性差,循环寿命短的新型锂硫电池体系[25-26].聚合物衍生锂硫电池体系相对于传统锂硫体系,以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是简单的物理负载(多孔碳载硫),故有着极大的应用前景[27]. ...
Asymmetric allyl-activation of organosulfides for high-energy reversible redox flow batteries
1
2019
... 虽然锂硫电池具有超高的理论能量密度,但在实际应用中却因固有的电导率低、多硫化物易穿梭等不可忽视的问题,导致其可实现的容量并不高.另外在循环过程中的正极材料储锂能力快速下降,循环寿命短,限制了锂硫电池实现商业化[24].因此,亟待开发出一种能有效提升活性硫的利用率,真正解决传统锂硫电池体系稳定性差,循环寿命短的新型锂硫电池体系[25-26].聚合物衍生锂硫电池体系相对于传统锂硫体系,以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是简单的物理负载(多孔碳载硫),故有着极大的应用前景[27]. ...
Synthesis of nitrogen-doped mesoporous carbon spheres with extra-large pores through assembly of diblock copolymer micelles
1
2015
... 虽然锂硫电池具有超高的理论能量密度,但在实际应用中却因固有的电导率低、多硫化物易穿梭等不可忽视的问题,导致其可实现的容量并不高.另外在循环过程中的正极材料储锂能力快速下降,循环寿命短,限制了锂硫电池实现商业化[24].因此,亟待开发出一种能有效提升活性硫的利用率,真正解决传统锂硫电池体系稳定性差,循环寿命短的新型锂硫电池体系[25-26].聚合物衍生锂硫电池体系相对于传统锂硫体系,以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是简单的物理负载(多孔碳载硫),故有着极大的应用前景[27]. ...
Double-shelled NiO-NiCo2O4 heterostructure@carbon hollow nanocages as an efficient sulfur host for advanced lithium-sulfur batteries
1
2018
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
Sulfurized polyacrylonitrile cathodes with high compatibility in both ether and carbonate electrolytes for ultrastable lithium-sulfur batteries
2
2019
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
... 虽然主链上的共轭并苯结构能一定程度改善反应体系的电导率,但受限于硫(5×10-30 S/cm)和多硫化锂(10-13 S/cm)的本征绝缘性,S@PAN仍存在反应动力学缓慢的问题.基于此,科研工作者们致力于通过掺入微量高电导率元素(Fe、Co、Ni、Se、Te等),设计新颖的形貌结构的和构筑连续的导电网络来加快体系的反应动力学.例如,Xie等[41]利用硒的高电导率(1×10-3 S/cm)和超高的体积容量(3253 mA·h/cm3),构筑了SexSPAN(x=0.06、0.09、0.14)复合材料作为锂硫电池正极[图4(a)].硒和硫是同主族元素,在提升体系反应动力学的同时,也能和锂离子反应贡献部分容量.同时,该文献认为S@PAN的储锂过程是“准固态反应”.在锂化过程中,硫链断裂形成多硫化锂(Li2Sn,1≤n≤4).当2≤n≤4,随着链状硫的变短,多硫化锂的溶解性下降,但仍有少部分可溶解在醚类电解液中产生穿梭效应,造成活性硫的流失,最终实际容量远低于理论容量.硒元素的引入加快了反应动力学,加快了Li2Sn的转化速率,最大程度上避免了活性硫的损失.因此,目标产物不论在醚类或酯类电解液中,均表现出优异的储锂性能:在0.2 A/g下能提供1300 mA·h/g的可逆容量(84%的活性物质利用率),在10 A/g的超高电流密度下,仍能保持900 mA·h/g的可逆容量,经800圈长循环,每圈的容量衰减低至0.029%.Liu等[29]将SPAN和碳纳米管混合,通过静电纺丝技术构筑了三维导电网络,加快电子的传输和离子的扩散,减缓了锂化/去锂化过程中的体积膨胀[图4(b)].同时证明PAN骨架中的π-π共轭键具有较强的键合作用,使得短链硫(—Sx—,x≤3)在醚类或者酯类电解液中均能发生可逆的氧化还原反应.基于此,SPAN/CNT-12在800 mA/g下对应着高达1100 mA·h/g的可逆容量.除此之外,主链中的“C=N”有利于提升反应体系的电导率,加快反应动力学.随后通过DFT理论计算,分析了S@PAN的储锂机理. ...
Trisulfide-bond acenes for organic batteries
2
2019
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
... 基于苯固有的共轭大π键有利于提升锂硫体系的电导率,含苯小分子有机硫电极也吸引了越来越多的关注.Preefer等[55]致力于研究苯环上含6个硫原子的交联有机物作为锂硫电池正极材料[图7(c)].每一个活性单元(C6S6)的理论比容量为609 mA·h/g,但是,由于硫的绝缘性导致活性单元较低的利用率,仅仅表现出150 mA·h/g的储锂性能.Long等[30]于2019年分别以四并苯和五并苯为原料,和单质硫混合,采用分段式加热的方法,分别合成了单晶四硫并苯(TTT)和单晶六硫五并苯(HTP),并作为锂硫电池正极.通过电化学测试和DFT理论计算,发现其反应机理分为两段式储能[图7(d)],分别在1.41/1.95 V和1.87/2.27 V处表现处两个充放电平台,在0.1 C下,其储锂容量分别为257和265 mA·h/g. ...
锂电池正极材料有机多硫化物的展望
1
2002
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
锂电池正极材料有机多硫化物的展望
1
2002
... 众所周知,单质硫较低的电导率(5×10-30 S/cm),循环过程中长链多硫化物的易穿梭性和锂负极的不稳定性,导致传统的锂硫电池很难实现商业化[5, 28].硫代聚丙烯腈体系(S@PAN)凭借其侧链中/短链硫(—C—Sx—C—,1≤x≤4)的难/不溶性和主链共轭导电结构,很好地解决上述问题[29].当单质硫(S8)和聚丙烯腈(PAN)在280~300 ℃下经过3~6 h退火处理时,PAN中的“—CN”在高温下易断键成环,同时升华硫作为一种良好的脱氢剂,易使中/短链硫与腈类聚合物环合形成复合材料,故而通过一步热处理法将共轭导电主链和活性有机硫支链整合到复合材料中,实现“主链导电,侧链储能”的目的[30-31]. ...
A novel conductive polymer-sulfur composite cathode material for rechargeable lithium batteries
1
2002
... 传统的锂硫电池设计主要集中于“微孔结构的物理限域”和“极性位点的化学吸附”两大思路,而S@PAN受益于其侧链上中/短链硫片段,在储锂过程中,不存在长链可溶性多硫化物的形成.早在2002年,Wang等[32-33]首次提出了S@PAN可作为可充电式锂电池正极,并具有优异的导电性和快速的离子传输速率的特点,相对于传统的锂硫正极表现出更优异的倍率性能,该文并未对其储锂机理进行深入分析.近年来,随着表征技术的飞速发展,S@PAN再次引起了科研工作者的关注,主要集中于对其具体结构和储锂机理的分析. ...
Sulfur composite cathode materials for rechargeable lithium batteries
1
2003
... 传统的锂硫电池设计主要集中于“微孔结构的物理限域”和“极性位点的化学吸附”两大思路,而S@PAN受益于其侧链上中/短链硫片段,在储锂过程中,不存在长链可溶性多硫化物的形成.早在2002年,Wang等[32-33]首次提出了S@PAN可作为可充电式锂电池正极,并具有优异的导电性和快速的离子传输速率的特点,相对于传统的锂硫正极表现出更优异的倍率性能,该文并未对其储锂机理进行深入分析.近年来,随着表征技术的飞速发展,S@PAN再次引起了科研工作者的关注,主要集中于对其具体结构和储锂机理的分析. ...
Structure-related electrochemistry of sulfur-poly(acrylonitrile) composite cathode materials for rechargeable lithium batteries
1
2011
... 针对于S@PAN的具体结构,科研工作者展开了积极的探讨.2011年,Buchmeiser等[34]报道S@PAN的结构如图2(a)所示,通过傅里叶-红外光谱(FT-IR),X-射线光电子能谱(XPS)和质谱测试(GC-MS)的佐证,认为短链硫(—C—Sx—C—,1≤x≤3)和 PAN 骨架在高温下同时进行脱氢和环化作用.Zhang等[35]于2014年通过元素分析和热重-质谱法(TG-MS)得到S@PAN的结构如图2(b)和2(c)所示,经计算,认为图2(a)中得到的结构不符合测定的C/H元素比.同时证明中/短链硫(—C—Sx—C—)的平均链长为x=3.37(n+2),且x的最大值不超过4.也就意味着在储锂过程中,避免了长链多硫化物的形成,因此能有效地改善传统锂硫体系循环稳定性差的问题.随后,Archer等[36]于2015年报道S@PAN的结构如图2(d)所示,在200 ℃以上腈键会断裂,形成具有π-π共轭、高导电性的并苯主链.其侧链由短链硫(—Sx—,x≤2)和中链硫(—Sx—,3≤x≤4)组成.Ming等[37]于2018年通过将13C固态核磁谱和量子化学模拟相结合,认为S@PAN的结构如图2(e)所示,并证明其S—S键首圈的断裂是不可逆的.随着表征技术的发展和科研工作的不断深入,科研工作者们对S@PAN结构的认识已趋于一致.但是,S@PAN在储锂过程中活性侧链片段(—Sx—)与主链导电聚合物(PAN)之间的相互作用仍然存在着争论. ...
Understanding of sulfurized polyacrylonitrile for superior performance lithium/sulfur battery
2
2014
... 针对于S@PAN的具体结构,科研工作者展开了积极的探讨.2011年,Buchmeiser等[34]报道S@PAN的结构如图2(a)所示,通过傅里叶-红外光谱(FT-IR),X-射线光电子能谱(XPS)和质谱测试(GC-MS)的佐证,认为短链硫(—C—Sx—C—,1≤x≤3)和 PAN 骨架在高温下同时进行脱氢和环化作用.Zhang等[35]于2014年通过元素分析和热重-质谱法(TG-MS)得到S@PAN的结构如图2(b)和2(c)所示,经计算,认为图2(a)中得到的结构不符合测定的C/H元素比.同时证明中/短链硫(—C—Sx—C—)的平均链长为x=3.37(n+2),且x的最大值不超过4.也就意味着在储锂过程中,避免了长链多硫化物的形成,因此能有效地改善传统锂硫体系循环稳定性差的问题.随后,Archer等[36]于2015年报道S@PAN的结构如图2(d)所示,在200 ℃以上腈键会断裂,形成具有π-π共轭、高导电性的并苯主链.其侧链由短链硫(—Sx—,x≤2)和中链硫(—Sx—,3≤x≤4)组成.Ming等[37]于2018年通过将13C固态核磁谱和量子化学模拟相结合,认为S@PAN的结构如图2(e)所示,并证明其S—S键首圈的断裂是不可逆的.随着表征技术的发展和科研工作的不断深入,科研工作者们对S@PAN结构的认识已趋于一致.但是,S@PAN在储锂过程中活性侧链片段(—Sx—)与主链导电聚合物(PAN)之间的相互作用仍然存在着争论. ...
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
Metal-sulfur battery cathodes based on pan-sulfur composites
2
2015
... 针对于S@PAN的具体结构,科研工作者展开了积极的探讨.2011年,Buchmeiser等[34]报道S@PAN的结构如图2(a)所示,通过傅里叶-红外光谱(FT-IR),X-射线光电子能谱(XPS)和质谱测试(GC-MS)的佐证,认为短链硫(—C—Sx—C—,1≤x≤3)和 PAN 骨架在高温下同时进行脱氢和环化作用.Zhang等[35]于2014年通过元素分析和热重-质谱法(TG-MS)得到S@PAN的结构如图2(b)和2(c)所示,经计算,认为图2(a)中得到的结构不符合测定的C/H元素比.同时证明中/短链硫(—C—Sx—C—)的平均链长为x=3.37(n+2),且x的最大值不超过4.也就意味着在储锂过程中,避免了长链多硫化物的形成,因此能有效地改善传统锂硫体系循环稳定性差的问题.随后,Archer等[36]于2015年报道S@PAN的结构如图2(d)所示,在200 ℃以上腈键会断裂,形成具有π-π共轭、高导电性的并苯主链.其侧链由短链硫(—Sx—,x≤2)和中链硫(—Sx—,3≤x≤4)组成.Ming等[37]于2018年通过将13C固态核磁谱和量子化学模拟相结合,认为S@PAN的结构如图2(e)所示,并证明其S—S键首圈的断裂是不可逆的.随着表征技术的发展和科研工作的不断深入,科研工作者们对S@PAN结构的认识已趋于一致.但是,S@PAN在储锂过程中活性侧链片段(—Sx—)与主链导电聚合物(PAN)之间的相互作用仍然存在着争论. ...
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
Recognizing the mechanism of sulfurized polyacrylonitrile cathode materials for Li-S batteries and beyond in Al-S batteries
2
2018
... 针对于S@PAN的具体结构,科研工作者展开了积极的探讨.2011年,Buchmeiser等[34]报道S@PAN的结构如图2(a)所示,通过傅里叶-红外光谱(FT-IR),X-射线光电子能谱(XPS)和质谱测试(GC-MS)的佐证,认为短链硫(—C—Sx—C—,1≤x≤3)和 PAN 骨架在高温下同时进行脱氢和环化作用.Zhang等[35]于2014年通过元素分析和热重-质谱法(TG-MS)得到S@PAN的结构如图2(b)和2(c)所示,经计算,认为图2(a)中得到的结构不符合测定的C/H元素比.同时证明中/短链硫(—C—Sx—C—)的平均链长为x=3.37(n+2),且x的最大值不超过4.也就意味着在储锂过程中,避免了长链多硫化物的形成,因此能有效地改善传统锂硫体系循环稳定性差的问题.随后,Archer等[36]于2015年报道S@PAN的结构如图2(d)所示,在200 ℃以上腈键会断裂,形成具有π-π共轭、高导电性的并苯主链.其侧链由短链硫(—Sx—,x≤2)和中链硫(—Sx—,3≤x≤4)组成.Ming等[37]于2018年通过将13C固态核磁谱和量子化学模拟相结合,认为S@PAN的结构如图2(e)所示,并证明其S—S键首圈的断裂是不可逆的.随着表征技术的发展和科研工作的不断深入,科研工作者们对S@PAN结构的认识已趋于一致.但是,S@PAN在储锂过程中活性侧链片段(—Sx—)与主链导电聚合物(PAN)之间的相互作用仍然存在着争论. ...
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
Lithium storage in conductive sulfur-containing polymers
1
2004
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
A new insight into the lithium storage mechanism of sulfurized polyacrylonitrile with no soluble intermediates
2
2018
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
... [39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
Organic polymer material with a multi-electron process redox reaction: Towards ultra-high reversible lithium storage capacity
1
2013
... 迄今为止,对S@PAN储锂机制的分析仍然存在许多争论.Wen等[38]早在2004年对其储锂机理进行了分析,提出在放电过程中“C—S”键不会断裂,“S—S”键在放电过程中断裂形成“Li—S—C”结构且该过程具有高度可逆性.除此之外,S@PAN中大量的微孔结构对锂离子产生物理吸附.但是,根据大量的文献报道,证明S@PAN并不存在微孔结构[39].Wang等[40]于2013年认为在首次放电过程中,“C—S”及“S—S”键断裂和锂离子反应生成Li2S[图3(a)];而在首次充电过程中形成的是元素硫而不是“C—S”及“S—S”键,随后硫化锂和分布在吡啶骨架中的元素硫之间发生可逆的化学转化,同时吡啶骨架之中的“C=C”,“C=N”键也能存储少量锂离子,因此其理论比容量高于传统的锂硫体系(1675 mA·h/g).该解释明显是不科学的,首先未考虑到中链硫(—Sx—,x=3、4)的存在,同时,假如是元素硫作为活性位点参与反应,应该分别出现高电位和低电位两个放电平台,这与实际充放电曲线相悖.随后,Zhang等[35]和Archer等[36]分别于2014、2015年提出了“C—S”和“S—S”键能够断裂和锂反应生成Li2S,随后Li2S能在充电过程中被氧化重新生成“C—S”和“S—S”键,但是该观点并不能很好地解释S@PAN体系的高理论容量.近年来,随着表征手段的逐渐完善,对于S@PAN体系的储锂机理研究也取得了显著的进展.Yang等[39]于2018年通过异位固态核磁共振波谱(SS-NMR)探究活性物质在充放电过程中的结构变化,同时通过异位X-射线光电子能谱探究不同电位下硫元素的存在状态[图3(b)].结果显示,在锂化过程中,“S—S”键的峰强逐渐变弱,在放电结束时消失,同时出现了Li2S的信号;而在去锂化过程中,Li2S的信号逐渐消失而“S—S”的信号重新出现,故储锂机制归因于“S—S”键和Li2S之间的可逆转化.除此之外,SS-NMR结果显示在锂化过程中“C=N”和“C=C”强度下降,同时出现了两组新峰“Li-C-N-Li”,“Li-C-C-Li”,且在去锂化的过程中“C=N”和“C=C”能够复原,说明“C=N”和“C=C”能够储存少量的锂,故其理论容量高于传统C/S体系.2018年,Ming等[37]基于S@PAN体系超高的稳定性(98.7%,2000圈),认为首圈“S—S”键断裂形成硫自由基,随后和吡啶环形成含硫自由基电子离域,在锂化/去锂化过程中,该共轭结构和锂发生可逆的反应产生电子转移,形成离子配位键,最终实现硫自由基和离子配位键之间的可逆转化[图3(c)].值得注意的是,该工作将7Li固态核磁共振和量子化学模拟相结合,证明Li+能和S,N周围的非活性区域结合通过可逆的锂耦合电子传递过程形成离子配位键,这是S@PAN体系具有优异循环稳定性的根本原因,而不是通常所认知的多硫化锂中间产物的形成.除此之外,电子自旋共振光谱的测试结果也验证了这一说法.随着表征手段的迅猛发展,科研工作者们对S@PAN体系储锂机理的认知变得更加科学和完善. ...
Ether-compatible sulfurized polyacrylonitrile cathode with excellent performance enabled by fast kinetics via selenium doping
1
2019
... 虽然主链上的共轭并苯结构能一定程度改善反应体系的电导率,但受限于硫(5×10-30 S/cm)和多硫化锂(10-13 S/cm)的本征绝缘性,S@PAN仍存在反应动力学缓慢的问题.基于此,科研工作者们致力于通过掺入微量高电导率元素(Fe、Co、Ni、Se、Te等),设计新颖的形貌结构的和构筑连续的导电网络来加快体系的反应动力学.例如,Xie等[41]利用硒的高电导率(1×10-3 S/cm)和超高的体积容量(3253 mA·h/cm3),构筑了SexSPAN(x=0.06、0.09、0.14)复合材料作为锂硫电池正极[图4(a)].硒和硫是同主族元素,在提升体系反应动力学的同时,也能和锂离子反应贡献部分容量.同时,该文献认为S@PAN的储锂过程是“准固态反应”.在锂化过程中,硫链断裂形成多硫化锂(Li2Sn,1≤n≤4).当2≤n≤4,随着链状硫的变短,多硫化锂的溶解性下降,但仍有少部分可溶解在醚类电解液中产生穿梭效应,造成活性硫的流失,最终实际容量远低于理论容量.硒元素的引入加快了反应动力学,加快了Li2Sn的转化速率,最大程度上避免了活性硫的损失.因此,目标产物不论在醚类或酯类电解液中,均表现出优异的储锂性能:在0.2 A/g下能提供1300 mA·h/g的可逆容量(84%的活性物质利用率),在10 A/g的超高电流密度下,仍能保持900 mA·h/g的可逆容量,经800圈长循环,每圈的容量衰减低至0.029%.Liu等[29]将SPAN和碳纳米管混合,通过静电纺丝技术构筑了三维导电网络,加快电子的传输和离子的扩散,减缓了锂化/去锂化过程中的体积膨胀[图4(b)].同时证明PAN骨架中的π-π共轭键具有较强的键合作用,使得短链硫(—Sx—,x≤3)在醚类或者酯类电解液中均能发生可逆的氧化还原反应.基于此,SPAN/CNT-12在800 mA/g下对应着高达1100 mA·h/g的可逆容量.除此之外,主链中的“C=N”有利于提升反应体系的电导率,加快反应动力学.随后通过DFT理论计算,分析了S@PAN的储锂机理. ...
The use of elemental sulfur as an alternative feedstock for polymeric materials
1
2013
... Pyun等[42]首次利用共聚法,将1,3-二异戊基苯(DIB)在185 ℃下浸在熔融态的单质硫中,进而制备了聚硫代二异丙烯基苯[poly(S-r-DIB)]材料[图5(a)].值得注意的是,该温度能同时满足1,3-二异戊基苯的不饱和双键裂解和硫单质的熔融,故不使用任何有机溶剂,也能一步实现目标产物的制备.与此同时,该体系首次采用共聚法将硫单质经过有机环合掺入可加工高分子材料中,利用压印光刻技术,调控有机材料DIB(10%~50%,质量分数)的质量分数来实现不同载硫量的poly(S-r-DIB)薄膜.将其作为锂硫电池正极,经过100圈循环后,其可逆比容量保持在823 mA·h/g,其储锂机理和传统锂硫电池基本一致. ...
Sulfur-rich polymeric materials with semi-interpenetrating network structure as a novel lithium-sulfur cathode
1
2014
... 随后,Meng等[43]利用1,3-二乙炔基苯(DEB)合成了更高含硫量的C—S共聚物,目标产物是由链状聚合物相互贯穿而形成牢笼形网络[图5(b)].核磁结果显示,高温处理后的DEB的共价炔键(C≡C)分解形成共价单键(C—C).目标材料表现出超高的首次放电容量(0.1 C下1143 mA·h/g),在1 C的电流密度下循环500圈,其容量保持率仍为初始值的70%.其较高的容量归因于较高的载硫量和较高的硫利用率,比起之前的研究更具有实用价值. ...
Improving the charge conductance of elemental sulfur via tandem inverse vulcanization and electropolymerization
1
2015
... 基于先前的DIB在185 ℃下环硫的工作,Pyun等[44]又开发出了一种新型的化学功能化法来实现聚合物和高载量的单质硫的共聚[图5(c)].首先将硫单质,苯乙烯的衍生物和3,4-烷基二氧噻吩(ProDOT-Sty)混合均匀并加热至180 ℃,在不饱和双键断裂的同时,单质硫也开环并进而连接到有机物链上,随后加入DIB经“逆硫化”过程形成聚合物主链,将若干个ProDOT-Sty片段串联起来.最后,采用电聚合的方法实现共轭性高含硫共聚物的制备,该复合材料能有效地改善锂硫电池导电性差的问题,有望用于高性能锂硫电池. ...
Copolymerization of polythiophene and sulfur to improve the electrochemical performance in lithium-sulfur batteries
1
2015
... Zentel等[45]首次将聚3-己基噻吩(P3HT)与单质硫混合,利用格氏复分解反应,在170 ℃下开环制备了具有C—S活性位点的复合材料(S-P3HT)并详细的表征通过共价键连接的S-P3HT.将目标材料作为锂硫电池正极,在0.5 C经过100圈的循环, ...
Synthesis of three-dimensionally interconnected sulfur-rich polymers for cathode materials of high-rate lithium-sulfur batteries
1
2015
... 基于上述反应机理,Park等[46]于2015年首次以多孔三聚硫氰酸晶体(TTCA)为反应单体,通过软模板法来调控有机硫复合物的形貌结构.值得注意的是,TTCA中的胺基在循环过程中能促进锂离子的扩散,进而解决了传统锂硫体系固有的导电性差的问题.具体的反应机理如图6(a)所示:①通过改变结晶溶剂,在TTCA分子间形成“N—H···S”氢键,在TTCA分子和溶剂之间形成“N—H···O=C”氢键,进而合成一系列TTCA共晶;②通过简单的加热处理去除晶体中的溶剂,形成多孔TTCA框架;③将硫单质在TTCA框架中浸渍;④将混合物在高温下开环加成.通过TGA证明材料中的硫以链状形式成功锚定在有机框架之中,而不是单纯的附着在有机物上.作为锂硫电池正极,在0.1 C下其可逆比容量高达1210 mA·h/g,当电流密度提升到5 C时,仍能保持730 mA·h/g的可逆比容量.经450圈长循环,其容量保持率为初始容量的83%. ...
Rational sulfur cathode design for lithium-sulfur batteries: Sulfur-embedded benzoxazine polymers
1
2016
... 随后,Choi等[47]于2016年采用一锅法合成了含硫聚苯并噁嗪(S-BOP),其含硫量高达72%[图6(b)].将S-BOP和单质硫混合,在高温经开环聚合得到目标富硫聚合物.其中,链状硫与S-BOP在高温下失去氢的硫醇自由基相结合,使得活性链状硫均匀地分布在整个聚合物中.作为锂硫电池正极,该体系在具有高的硫含量(72%)的同时,也具有优异的循环性能:S-BOP电极的初始库仑效率(ICE)高达96.6%,经过1000次长循环后,其容量保持率仍能达到初始值的92.7%.S-BOP的优异和循环性能揭示了有机硫正极材料的可行性,基于单体设计思想,进而有效调控硫聚合物的储锂性能. ...
Highly crosslinked organosulfur copolymer nanosheets with abundant mesopores as cathode materials for efficient lithium-sulfur batteries
1
2018
... 除此之外,Chen等[48]于2018年通过逆硫化作用将富硫链固定在席夫碱上获得硫脲醛树脂共聚物 [cp(S-TAR)]介孔纳米片[图6(c)].“C=S”基在高温下断键形成活性自由基,该自由基和硫链片段相结合,使得侧链上具有链状硫活性片段.将cp(S-TAR)作为锂硫电池正极,不仅为锂离子的扩散提供了丰富的孔道结构,同时基于富硫侧链和硫脲醛树脂之间强烈的共价键作用,较大程度地约束了多硫化物的溶解.在1 C下经过500圈长循环,其平均每圈的容量衰减仅为0.045%.该策略显示了高交联有机硫共聚物作为低成本、高能量密度锂电池的高性能聚合物正极材料的巨大潜力. ...
Ionic conductivity of organosulfur melts for advanced storage electrodes
1
1989
... 早在1988年,Visco 等[49]首次提出了有机小分子硫的概念并将二苯二硫(DPDS)和二硫化四乙基秋兰姆(TEDE)用于电化学储锂过程中,认为其中活性位点“S—S”键在锂化/去锂化过程中会断裂/重组,但是由于其较低的容量和较差的倍率性能,相对于过渡金属氧化物并没有太大的竞争力.但是其理论比容量较高,科研工作者们设计了不同的载体来固定小分子硫,进而改善其循环稳定性. ...
Electrochemical properties of organic disulfide/thiolate redox couples
1
1989
... 对于最简单的S—S来说,在锂化过程中,结构单元可吸收2e-和2Li+,而化合物中所含活性“S—S”键的量决定了其储锂能力.DeJonghe等[50]利用旋转圆盘电极首次确定了“S—S”在储能过程所对应的氧化还原反应(R-S2-R+2e-+2Li+2R-SLi).随着硫链长的增加,Fu等[51]在2016年将实验和DFT理论计算相结合,确定了二甲基三硫醚(DMTS)的储锂机理(CH3SSSCH3+4e-+4Li+2CH3SLi+Li2S).随后,Fu等[52]在同年报道了二苯基硫醚(DPTS)的储锂方式(PhSSSPh+4e-+4Li+2PhSLi+Li2S).因此,小分子有机硫的反应机理可以总结成反应式:R-Sn-R+(2n-2)e-+(2n-2)Li+2R-SLi+(n-2)Li2S,n≥2.如图7(a)所示,R-Sx-R(2≤x≤6)的理论比容量随着活性位点占比的增加而增大[53]. ...
Organotrisulfide: A high capacity cathode material for rechargeable lithium batteries
1
2016
... 对于最简单的S—S来说,在锂化过程中,结构单元可吸收2e-和2Li+,而化合物中所含活性“S—S”键的量决定了其储锂能力.DeJonghe等[50]利用旋转圆盘电极首次确定了“S—S”在储能过程所对应的氧化还原反应(R-S2-R+2e-+2Li+2R-SLi).随着硫链长的增加,Fu等[51]在2016年将实验和DFT理论计算相结合,确定了二甲基三硫醚(DMTS)的储锂机理(CH3SSSCH3+4e-+4Li+2CH3SLi+Li2S).随后,Fu等[52]在同年报道了二苯基硫醚(DPTS)的储锂方式(PhSSSPh+4e-+4Li+2PhSLi+Li2S).因此,小分子有机硫的反应机理可以总结成反应式:R-Sn-R+(2n-2)e-+(2n-2)Li+2R-SLi+(n-2)Li2S,n≥2.如图7(a)所示,R-Sx-R(2≤x≤6)的理论比容量随着活性位点占比的增加而增大[53]. ...
Highly reversible diphenyl trisulfide catholyte for rechargeable lithium batteries
1
2016
... 对于最简单的S—S来说,在锂化过程中,结构单元可吸收2e-和2Li+,而化合物中所含活性“S—S”键的量决定了其储锂能力.DeJonghe等[50]利用旋转圆盘电极首次确定了“S—S”在储能过程所对应的氧化还原反应(R-S2-R+2e-+2Li+2R-SLi).随着硫链长的增加,Fu等[51]在2016年将实验和DFT理论计算相结合,确定了二甲基三硫醚(DMTS)的储锂机理(CH3SSSCH3+4e-+4Li+2CH3SLi+Li2S).随后,Fu等[52]在同年报道了二苯基硫醚(DPTS)的储锂方式(PhSSSPh+4e-+4Li+2PhSLi+Li2S).因此,小分子有机硫的反应机理可以总结成反应式:R-Sn-R+(2n-2)e-+(2n-2)Li+2R-SLi+(n-2)Li2S,n≥2.如图7(a)所示,R-Sx-R(2≤x≤6)的理论比容量随着活性位点占比的增加而增大[53]. ...
Organosulfides: An emerging class of cathode materials for rechargeable lithium batteries
1
2019
... 对于最简单的S—S来说,在锂化过程中,结构单元可吸收2e-和2Li+,而化合物中所含活性“S—S”键的量决定了其储锂能力.DeJonghe等[50]利用旋转圆盘电极首次确定了“S—S”在储能过程所对应的氧化还原反应(R-S2-R+2e-+2Li+2R-SLi).随着硫链长的增加,Fu等[51]在2016年将实验和DFT理论计算相结合,确定了二甲基三硫醚(DMTS)的储锂机理(CH3SSSCH3+4e-+4Li+2CH3SLi+Li2S).随后,Fu等[52]在同年报道了二苯基硫醚(DPTS)的储锂方式(PhSSSPh+4e-+4Li+2PhSLi+Li2S).因此,小分子有机硫的反应机理可以总结成反应式:R-Sn-R+(2n-2)e-+(2n-2)Li+2R-SLi+(n-2)Li2S,n≥2.如图7(a)所示,R-Sx-R(2≤x≤6)的理论比容量随着活性位点占比的增加而增大[53]. ...
The unique chemistry of thiuram polysulfides enables energy dense lithium batteries
1
2017
... 氮元素凭借较强的电负性(-3.04 V,vs.SHE),在锂硫电池体系类呈现路易斯酸的特征,对多硫化物有着较强的化学吸附作用.Fu等[54]于2017年报导将二硫化双亚戊基秋兰姆(PMTT)作为锂硫电池正极,在2.57,2.34和2.04 V出现3个稳定的平台[图7(b)].每个PMTT活性片段能存储6个锂离子,其理论比容量为418 mA·h/g.随后,通过DFT理论计算证明其锂化产物为LiPMDTC和Li2S,其中LiPMDTC+以离子状态存在时有利于电荷转移. ...
High sulfur content material with stable cycling in lithium-sulfur batteries
1
2017
... 基于苯固有的共轭大π键有利于提升锂硫体系的电导率,含苯小分子有机硫电极也吸引了越来越多的关注.Preefer等[55]致力于研究苯环上含6个硫原子的交联有机物作为锂硫电池正极材料[图7(c)].每一个活性单元(C6S6)的理论比容量为609 mA·h/g,但是,由于硫的绝缘性导致活性单元较低的利用率,仅仅表现出150 mA·h/g的储锂性能.Long等[30]于2019年分别以四并苯和五并苯为原料,和单质硫混合,采用分段式加热的方法,分别合成了单晶四硫并苯(TTT)和单晶六硫五并苯(HTP),并作为锂硫电池正极.通过电化学测试和DFT理论计算,发现其反应机理分为两段式储能[图7(d)],分别在1.41/1.95 V和1.87/2.27 V处表现处两个充放电平台,在0.1 C下,其储锂容量分别为257和265 mA·h/g. ...
Controllable chain-length for covalent sulfur-carbon materials enabling stable and high-capacity sodium storage
1
2019
... 随着电子设备的迅猛发展,构筑高能量密度,长服役时间,高安全性和低成本的电化学存储器件迫在眉睫.近年来,聚合物衍生硫正极凭借其较高的能量密度,优异的循环稳定性和较低的成本吸引了越来越多科研工作者的关注,相较于锂离子电池,同时具有降低成本和提升能量密度的优势;相较于传统的锂硫电池,虽然能量密度有所下降,但却解决了单质硫正极固有的循环性能差的问题.本文综述了关于聚合物衍生锂硫电极的发展和近期的工作,包括腈基衍生有机硫电极,不饱和烃基衍生有机硫电极,硫醇基衍生有机硫电极和小分子有机硫电极(图8),相对于传统锂硫体系,聚合物衍生锂硫电极以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是作为简单的载体,因而有着极大的潜力大幅度提升锂硫电池体系的稳定性.但是,其循环稳定性和活性硫的利用率仍需进一步的优化,因而距离实际应用还有着不小的差距.因此,未来的发展方向主要集中于以下四个方面:①设计新型的分子量分布较窄的不饱和有机物,并与硫进行高温环合,进一步抑制多硫化物的“穿梭效应”;②引入兼具高导电率和高催化效应的金属元素(Se、Te等),优化有机硫体系缓慢的反应动力学问题,促进离子扩散和电子传输,提高活性位点的利用率;③研究具有良好力学性能的不饱和聚合物环硫,适应锂离子在嵌入/脱嵌过程产生的形变,增加体系的循环稳定性;④优化电解质设计,保护锂金属负极,提升体系的安全性和在循环过程中的稳定性.除此之外,Ji等近年来实现了锂电到钠电的过度,将有机硫电极作为钠电正极,设计制备了不同碳硫链长度的硫碳材料[56],并系统地研究这些材料的表界面共价键变化与其储能行为之间的关系[57],证明了该类材料在钠电领域也具有应用潜力.进一步优化电解质,设计特殊的形貌结构,引入高电导元素,构筑导电网络是后续聚合物衍生锂硫正极研究的努力方向,也是走向市场的必经之路. ...
Surface-driven energy storage behavior of dual-heteroatoms functionalized carbon material
1
2019
... 随着电子设备的迅猛发展,构筑高能量密度,长服役时间,高安全性和低成本的电化学存储器件迫在眉睫.近年来,聚合物衍生硫正极凭借其较高的能量密度,优异的循环稳定性和较低的成本吸引了越来越多科研工作者的关注,相较于锂离子电池,同时具有降低成本和提升能量密度的优势;相较于传统的锂硫电池,虽然能量密度有所下降,但却解决了单质硫正极固有的循环性能差的问题.本文综述了关于聚合物衍生锂硫电极的发展和近期的工作,包括腈基衍生有机硫电极,不饱和烃基衍生有机硫电极,硫醇基衍生有机硫电极和小分子有机硫电极(图8),相对于传统锂硫体系,聚合物衍生锂硫电极以共价键的形式将整个/部分活性硫片段整合到正极材料中,而不是作为简单的载体,因而有着极大的潜力大幅度提升锂硫电池体系的稳定性.但是,其循环稳定性和活性硫的利用率仍需进一步的优化,因而距离实际应用还有着不小的差距.因此,未来的发展方向主要集中于以下四个方面:①设计新型的分子量分布较窄的不饱和有机物,并与硫进行高温环合,进一步抑制多硫化物的“穿梭效应”;②引入兼具高导电率和高催化效应的金属元素(Se、Te等),优化有机硫体系缓慢的反应动力学问题,促进离子扩散和电子传输,提高活性位点的利用率;③研究具有良好力学性能的不饱和聚合物环硫,适应锂离子在嵌入/脱嵌过程产生的形变,增加体系的循环稳定性;④优化电解质设计,保护锂金属负极,提升体系的安全性和在循环过程中的稳定性.除此之外,Ji等近年来实现了锂电到钠电的过度,将有机硫电极作为钠电正极,设计制备了不同碳硫链长度的硫碳材料[56],并系统地研究这些材料的表界面共价键变化与其储能行为之间的关系[57],证明了该类材料在钠电领域也具有应用潜力.进一步优化电解质,设计特殊的形貌结构,引入高电导元素,构筑导电网络是后续聚合物衍生锂硫正极研究的努力方向,也是走向市场的必经之路. ...











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}