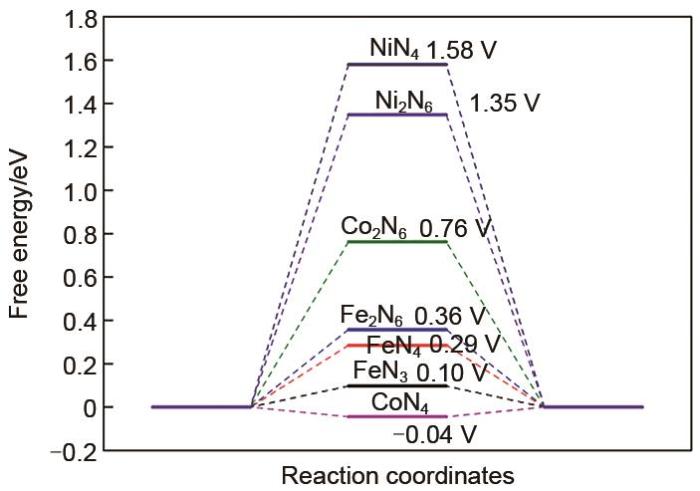
Electrocatalytic hydrogen evolution reaction (HER) is a promising hydrogen energy conversion method. To develop high-performance and low-cost hydrogen evolution electrocatalysts, single- and double-atom catalysts (SACs, DACs) with transition metals (e.g., Fe, Ni, and Co) as the active center and nitrogen-doped graphene (N-graphene) as the substrate are selected for HER utilizing density functional theory calculations. The selected catalytic materials exhibit exceptional stability against sintering. We then chose H adsorption energy as the descriptor for analyzing the HER activity, and the results demonstrate that the CoN4 site exhibits excellent HER activity over other candidates. In contrast, NiN4 and Ni2N6 sites display inferior HER activity. In addition, the electronic structures of the catalysts are systematically discussed to uncover the origin of catalytic activity. This work reveals that DACs have poor HER activity compared to SACs and DACs, while the SACs (e.g., CoN4, FeN3, and FeN4) show low overpotential in HER. Therefore, the SACs can substitute commercial precious metals catalysts (Pt/C) for HER catalysts.
Keywords:hydrogen evolution reaction
;
single-atom catalyst
;
electrocatalyst
;
density functional theory
ZHANG Shishi. Activity origin of single/double-atom catalyst for hydrogen evolution reaction[J]. Energy Storage Science and Technology, 2021, 10(6): 2008-2012
Fig. 1
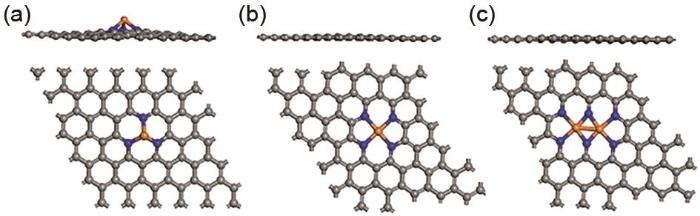
The side and top view of the geometry optimization structures of (a) FeN3, (b) FeN4, (c) Fe2N6-graphene. Cray, blue, orange balls represent C, N, Fe atoms, respectively
Fig. 2
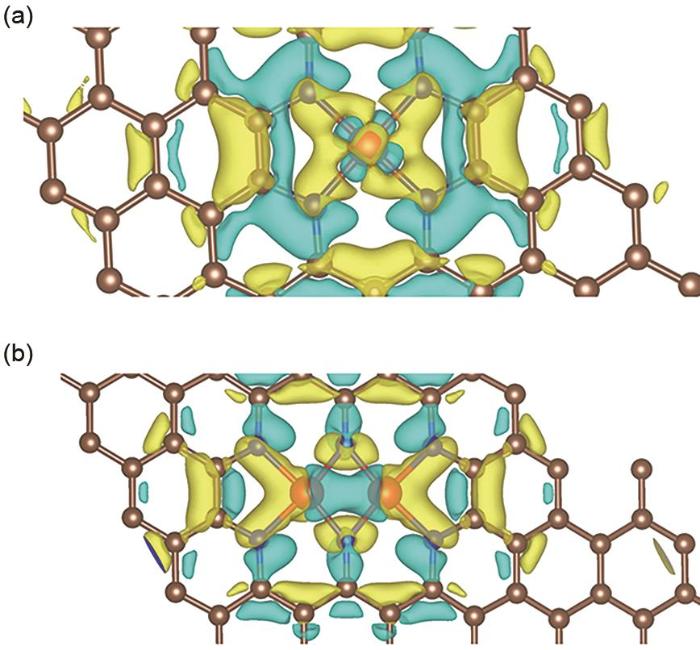
The differential charge density maps of (a) CoN4, (b) Co2N6, Brown, blue, red balls represent C, N, Co atoms, respectively, blue and yellow shadows characterize the charge loss and accumulation
Fig. 4
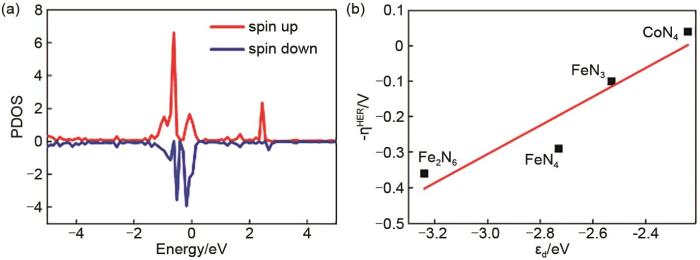
(a) Projected density of state (PDOS) projected on 3d orbitals of Co atom of CoN4. (b) The calculated linear relation of the ORR overpotential (-ηORR) as a function of d-band center (εd)values of Co atom
Fig. 5
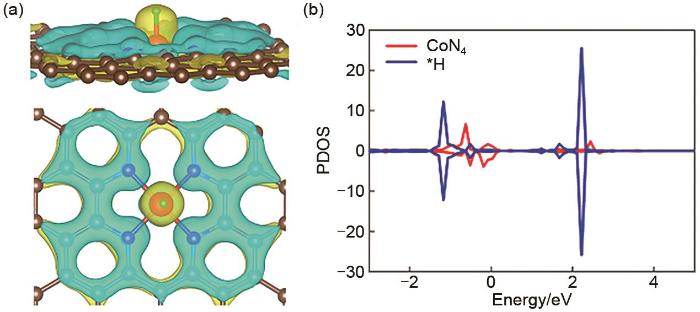
(a) The differential charge density maps where the brown, blue, red, green balls denote the C, N, Co, H atoms, respectively; (b) The projected density of state (PDOS) projected on 3d orbitals of Co atom and the 1s orbital of adsorbed H of CoN4-H
ZHANG H B, LIU G G, SHI L, et al. Single-atom catalysts: Emerging multifunctional materials in heterogeneous catalysis[J]. Advanced Energy Materials, 2018, 8(1): 1701343.
YANG S, KIM J, TAK Y J, et al. Single-atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions[J]. Angewandte Chemie International Edition, 2016, 55(6): 2058-2062.
XU H, WANG D, YANG P, et al. A theoretical study of atomically dispersed MN4/C (M=Fe or Mn) as a high-activity catalyst for the oxygen reduction reaction[J]. Physical Chemistry Chemical Physics, 2020, 22(48): 28297-28303.
ZHAO J, CHEN Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study[J]. Journal of the American Chemical Society, 2017, 139(36): 12480-12487.
WANG D W, LI Q, HAN C, et al. Single-atom ruthenium based catalyst for enhanced hydrogen evolution[J]. Applied Catalysis B: Environmental, 2019, 249: 91-97.
LIU X H, ZHENG L R, HAN C X, et al. Identifying the activity origin of a cobalt single-atom catalyst for hydrogen evolution using supervised learning[J]. Advanced Functional Materials, 2021, 31(18): 2100547.
CHENG N, STAMBULA S, WANG D, et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction[J]. Nature Communications, 2016, 7: 13638.
LEI C J, WANG Y, HOU Y, et al. Efficient alkaline hydrogen evolution on atomically dispersed Ni-Nx Species anchored porous carbon with embedded Ni nanoparticles by accelerating water dissociation kinetics[J]. Energy & Environmental Science, 2019, 12(1): 149-156.
CHEN Z W, YAN J M, JIANG Q. Single or double: Which is the altar of atomic catalysts for nitrogen reduction reaction?[J]. Small Methods, 2019, 3(6): 1800291.
LI Y C, LIU X F, ZHENG L R, et al. Preparation of Fe-N-C catalysts with FeNx (x=1, 3, 4) active sites and comparison of their activities for the oxygen reduction reaction and performances in proton exchange membrane fuel cells[J]. Journal of Materials Chemistry A, 2019, 7(45): 26147-26153.
NØRSKOV J K, BLIGAARD T, LOGADOTTIR A, et al. Trends in the exchange current for hydrogen evolution[J]. Journal of the Electrochemical Society, 2005, 152(3): J23.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}