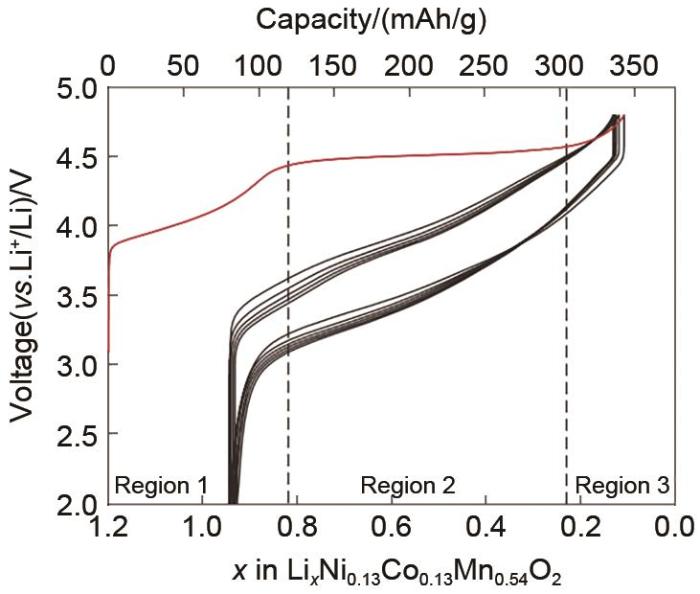
Layered Li-Mn-rich materials (LMR) are promising to be next generation cathodes for lithium-ion batteries due to their high specific capacity (>250 mAh/g) and low cost. It has been nearly 30 years since the discovery of LMR material, but it has never been commercialized for the following reasons: During the cycling process, Mn3+ migrates into Li sites makes the layered structure transform to spinel structure, resulting in a serious discharge voltage decay, which causing serious energy loss and bringing great challenges to battery management. The low electronic conductivity of Li2MnO3 makes LMR material have poor rate capability and lower electrode density results in lower volume energy density of LMR. In addition, the LMR materials need to be at high voltage (>4.55 V) to show high capacity, but the electrolyte at high voltage is easy to oxidize and decompose, accompanied by the release of lattice oxygen into O2, the above problems seriously affected LMR commercialization process. Based on the research and development results of LMR materials over the years, this paper reviews the research progress of LMR materials in the understanding of charge and discharge mechanism, precursor process route selection, modification effect and mechanism of bulk doping, surface coating, liquid and gas phase post-treatment, and the design of new special structures such as O2/O3 composite structure and single crystal structure. In addition, the future development direction and commercial prospect of LMR materials are prospected to help the industrial development of LMR materials.
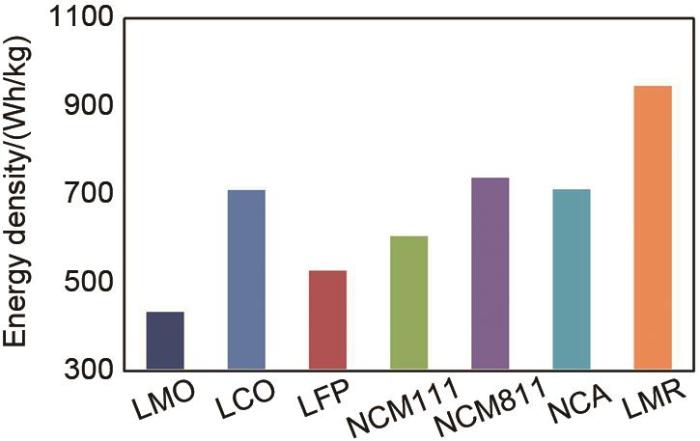
目前,传统的锂离子正极材料主要包括层状LiCoO2(LCO)、层状多元LiNi1-x-y Co x Mn y O2(NCM)和LiNi1-x-y Co x Al y O2(NCA)、橄榄石型结构LiFePO4(LFP)和尖晶石结构LiMn2O4(LMO)。LCO价格昂贵,主要用于3 C数码电子产品;多元NCM和NCA由于元素之间协同效应,循环性能明显提升,大规模应用于新能源动力电池,但其能量密度已不能满足人们对长续航的需求;LFP具有价格便宜、安全性能高和循环寿命长等优点,主要用于动力电池和储能领域,但其比容量较低,已接近理论值,难以取得突破;LMO价格低廉,容易发生锰的溶解和Jahn-Teller效应,导致高温循环性能变差,主要用于两轮车和电动工具。
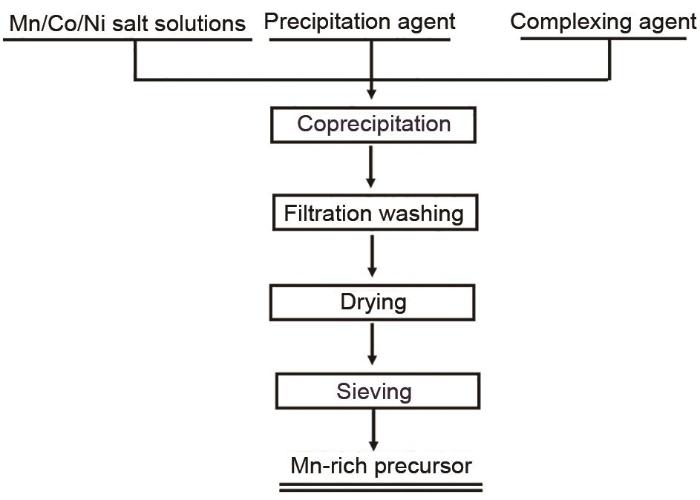
前驱体的形貌和微观结构对LMR正极材料的最终电化学性能具有非常重要的影响,常见的LMR前驱体合成方法有共沉淀法、溶胶-凝胶法和水热法等,但仅有共沉淀法适用于大规模应用。图3为富锂锰基前驱体共沉淀法制备工艺流程图,若沉淀剂采用碳酸钠,得到的LMR前驱体为碳酸盐前驱体Mn1-x-y Co x Ni y CO3,若沉淀剂采用氢氧化钠,得到的LMR前驱体为氢氧化物前驱体Mn1-x-y Co x Ni y (OH)2。
Fig. 3
Preparation process of LMR precursor by coprecipitation method
如图4所示,Mn1-x Ni x CO3和Mn1-x Ni x (OH)2前驱体的形貌结构具有明显的差异,Mn1-x Ni x CO3和Mn1-x Ni x (OH)2前驱体分别是由一次颗粒和片层结构组成,由于正极材料的形貌结构对前驱体具有一定的继承性,因此由Mn1-x Ni x CO3前驱体制备的正极材料LR-C具有更高的比表面积(约5 m2/g)和低的机械强度,在极片制作或循环过程中容易发生破碎,使得材料的循环性能变差。由Mn1-x Ni x (OH)2前驱体制备的正极材料LR-H具有更低的比表面积(约3 m2/g)和较好的机械强度。图4的充放电曲线表明与LR-H相比(249 mAh/g),LR-C具有略高的放电比容量(252 mAh/g),这是由于其更小的一次颗粒和较大的比表面积更有助于容量的发挥。但与LR-C循环性能对比结果表明LR-H具有更好的容量保持率和更低的电压降,循环80次的容量保持率从80%提高到93%,电压降从220 mV降低为140 mV。因此从商业化应用角度来看,氢氧化物前驱体更加适合。
图4
(a) Mn1-x Ni x CO3 和Mn1-x Ni x (OH)2 前驱体SEM图;LR-C和LR-H正极材料的 (b) 充放电曲线和 (c)~(d) 循环性能
Fig. 4
(a) SEM images of Mn1-x Ni x CO 3 and Mn1-x Ni x (OH)2 precursors; (b) The charge-discharge curves and (c)-(d) cycle life of LR-C and LR-H cathode materials
Fig. 5
(a) Active(010)planes and (b) rate capability of Li1.2Mn0.6Ni0.2O2 materials[7]; (c) Element distribution, (d) capacity and (e) average voltage cycle performance of Li1.2Mn0.6Ni0.2O2 materials[8]
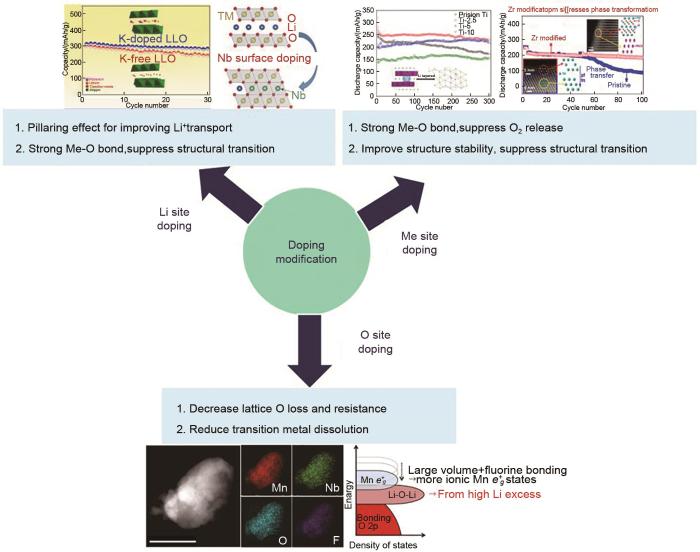
由于Na、K元素和Li位于同一主族,早先人们利用Na和K等低价元素对材料进行掺杂改性。Lim等[9]研究了不同Na含量掺杂对Li1.167-x Na x Ni0.18Mn0.548Co0.105O2(0≥x≥0.1)性能的影响,发现当x=0.05时,锂离子扩散系数从1.35×10-9 cm2/s提高到3.34×10-9 cm2/s,容量保持率从83%提高到92%,倍率性能也得到一定的改善。Li等[10]通过采用含有K元素的α-MnO2作为原料,经过原位合成得到K+掺杂的Li1.2Mn0.54Co0.13Ni0.13O2,并对K元素的作用进行了系统性研究,发现半径较大的K+可以降低锂层三空位的形成和锰的迁移,阻碍尖晶石相结构的形成,因此,该材料表现出很好的循环稳定性,循环110周后,容量保持率为85%。进一步研究发现如果Na+或K+在材料中均匀地掺杂,由于位阻效应的存在,使得材料不会从层状结构向尖晶石结构的转变,除非发生明显的晶格畸变。但在长时间循环过程中由于Jahn-Teller畸变会产生Mn3+,Mn3+再发生歧化反应生成Mn2+,Mn2+更倾向于四面体位点形成尖晶石相。因此,Na和K元素掺杂的材料最终还是会形成尖晶石相,只不过相比不掺杂的材料这一过程会被延缓而已。
即使相同的掺杂元素,采用不同的制备方法和工艺也有可能影响元素的掺杂位置和材料的性能。例如,前面提到的Ti4+掺杂在锂位,而日产汽车Yama moto等[16]采用喷雾热解法制备了Ti4+掺杂的Li1.5Ni0.25Mn0.75-x Ti x O2.5,通过TEM和EDX分析证明了Ti均匀地取代了Mn的位置,认为Ti掺杂可以抑制循环过程中Ni和Mn的溶出,因此提高了材料的循环性能。
Kim等[17]通过研究F掺杂的0.3Li2MnO3·0.7Li(Mn0.60Ni0.25Co0.15)O1.975F0.025,发现F掺杂能够形成更稳定的SEI层来提高Li+扩散速率,同时减少过渡金属的溶解和降低循环过程中的内阻。Lim等[18]通过研究F掺杂的Li1.2-x Mn0.54Ni0.13Co0.13O2-x F x (x=0.08),认为F取代部分O后可以缓解层状结构向尖晶石结构的转变,降低循环过程中的电压降,提高循环性能。因此,该材料经过100周循环后,容量保持率仍为95%,远高于未掺F材料的容量保持率(62%)。此外,F元素和其他金属元素共掺杂也成为了近年来的研究热点,凭借各自的优势互补作用,可以使改性效果更加明显。例如,加州大学伯克利分校的Lee等[19]采用球磨机械化学法合成了共掺杂的岩盐相Li2Mn2/3Nb1/3O2F和Li2Mn1/2Ti1/2O2F两种材料,发现共掺杂可以实现Mn2+/Mn4+的可逆氧化还原反应,因此减少了氧的氧化还原活性,使材料结构稳定。其中,Li2Mn2/3Nb1/3O2F表现出高的容量(>300 mAh/g)和能量密度(约1000 Wh/kg)。利用F掺杂到O位可以减小晶格氧的流失,缓解结构的转变,但由于富锂锰基的高容量是由于氧发生了氧化还原反应,因此,F掺杂可能会导致容量有一定降低。
在富锂锰基材料表面会涉及很多重要的反应,如电解液的分解、氧气逸出、过渡金属溶解等。近年来,研究发现对材料表面进行包覆处理可以有效地提高LMR材料的表面结构稳定性,降低电压降和提升循环、倍率性能。Wu等[20]发现Al2O3包覆可以明显地降低首次不可逆容量损失和提高放电容量,并且表现出优异的循环稳定性,作者认为Al2O3包覆抑制了材料表面和电解质之间的副反应。Zhao等[21]采用原子层沉积法在Li(Li0.2Mn0.54Ni0.13Co0.13)O2的表面进行了ZrO2和ZnO包覆,并研究了包覆层的厚度和组成对材料性能的影响,发现采用约10 nm LiCoO2和6层ZrO2原子层厚度的双壳层包覆时,材料表现出最高的放电容量(296.4 mAh/g)和优异的循环性能、倍率性能,作者认为该双层功能性包覆的协同效应能够降低循环过程中的电化学极化、结构变化和表面副反应。北京理工大学Wu等[22]通过研究MnO x 包覆的Li(Ni0.2Li0.2Mn0.6)O2,发现10%的MnO x (厚约20 nm)包覆量能够有效地改善材料的电化学性能,作者认为这是由于MnO x 包覆不仅能够保留材料层状结构内部的部分Li空位,本身还可以提供Li+嵌入位点。
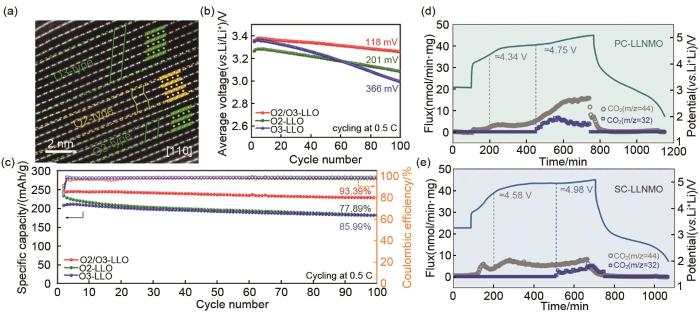
Fig. 8
(a) STEM image, (b) voltage and (c) capacity cycle performance of O2/O3 type LMR materials[33]; Charge-discharge curve and gas evolution comparison of (d) polycrystal and (e) single crystal LMR materials[35]
LUO K, ROBERTS M R, HAO R, et al. Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen[J]. Nature Chemistry, 2016, 8(7): 684-691.
HAN S J, XIA Y G, WEI Z, et al. A comparative study on the oxidation state of lattice oxygen among Li1.14Ni0.136Co0.136Mn0.544O2, Li2MnO3, LiNi0.5Co0.2Mn0.3O2 and LiCoO2 for the initial charge-discharge[J]. Journal of Materials Chemistry A, 2015, 3(22): 11930-11939.
SATHIYA M, RAMESHA K, ROUSSE G, et al. High performance Li2Ru1-yMnyO3 (0.2≤y≤0.8) cathode materials for rechargeable lithium-ion batteries: Their understanding[J]. Chemistry of Materials, 2013, 25(7): 1121-1131.
KOGA H, CROGUENNEC L, MÉNÉTRIER M, et al. Different oxygen redox participation for bulk and surface: A possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2[J]. Journal of Power Sources, 2013, 236: 250-258.
FREIRE M, KOSOVA N V, JORDY C, et al. A new active Li-Mn-O compound for high energy density Li-ion batteries[J]. Nature Materials, 2016, 15(2): 173-177.
CHEN L, SU Y, CHEN S, et al. Hierarchical Li1.2 Ni0.2 Mn0.6 O2 nanoplates with exposed{010}planes as high-performance cathode material for lithium-ion batteries[J]. Advanced Materials (Deerfield Beach, Fla), 2014, 26(39): 6756-6760.
ZHENG J M, GU M, GENC A, et al. Mitigating voltage fade in cathode materials by improving the atomic level uniformity of elemental distribution[J]. Nano Letters, 2014, 14(5): 2628-2635.
LIM S N, SEO J Y, JUNG D S, et al. Rate capability for Na-doped Li1.167Ni0.18Mn0.548Co0.105O2 cathode material and characterization of Li-ion diffusion using galvanostatic intermittent titration technique[J]. Journal of Alloys and Compounds, 2015, 623: 55-61.
LI Q, LI G S, FU C C, et al. K(+)-doped Li1.2Mn0.54Co0.13Ni0.13O2: A novel cathode material with an enhanced cycling stability for lithium-ion batteries[J]. ACS Applied Materials & Interfaces, 2014, 6(13): 10330-10341.
FENG X, GAO Y R, BEN L B, et al. Enhanced electrochemical performance of Ti-doped Li1.2Mn0.54Co0.13Ni0.13O2 for lithium-ion batteries[J]. Journal of Power Sources, 2016, 317: 74-80.
LIU S,LIU Z,SHEN X,et al. Surface doping to enhance structural integrity and performance of Li-rich layered oxide[J]. Advanced Energy Materials 2018,8: doi: 10.1002/aenm.201802105.
PARK S H, SUN Y K. Synthesis and electrochemical properties of layered Li[Li0.15Ni(0.275-x/2)AlxMn(0.575-x/2)]O2 materials prepared by sol-gel method[J]. Journal of Power Sources, 2003, 119/120/121: 161-165.
JIN X, XU Q J, LIU H M, et al. Excellent rate capability of Mg doped Li[Li0.2Ni0.13Co0.13Mn0.54]O2 cathode material for lithium-ion battery[J]. Electrochimica Acta, 2014, 136: 19-26.
LI X, ZHANG K, MITLIN D, et al. Fundamental insight into Zr modification of Li-and Mn-rich cathodes: combined transmission electron microscopy and electrochemical impedance spectroscopy study[J]. Chemistry of Materials, 2018, 30(8): 2566-2573.
YAMAMOTO S, NOGUCHI H, ZHAO W W. Improvement of cycling performance in Ti substituted 0.5Li2MnO3-0.5LiNi0.5Mn0.5O2 through suppressing metal dissolution[J]. Journal of Power Sources, 2015, 278: 76-86.
KIM S M, JIN B S, LEE S M, et al. Effects of the fluorine-substitution and acid treatment on the electrochemical performances of 0.3Li2MnO3·0.7LiMn0.60Ni0.25Co0.15O2 cathode material for Li-ion battery[J]. Electrochimica Acta, 2015, 171: 35-41.
LIM S N, SEO J Y, JUNG D S, et al. The crystal structure and electrochemical performance of Li1.167Mn0.548Ni0.18Co0.105O2 composite cathodes doped and co-doped with Mg and F[J]. Journal of Electroanalytical Chemistry, 2015, 740: 88-94.
WU Y, MANTHIRAM A. High capacity, surface-modified layered Li[Li(1-x)/3Mn(2-x)/3Nix/3Cox/3]O2 cathodes with low irreversible capacity loss[J]. Electrochemical and Solid-State Letters, 2006,9(5):A221-A224.
ZHAO J Q, AZIZ S, WANG Y. Hierarchical functional layers on high-capacity lithium-excess cathodes for superior lithium ion batteries[J]. Journal of Power Sources, 2014, 247: 95-104.
WU F, LI N, SU Y F, et al. Can surface modification be more effective to enhance the electrochemical performance of lithium rich materials? [J]. Journal of Materials Chemistry, 2012, 22(4): 1489-1497.
ZHENG J, GU M, XIAO J, et al. Functioning mechanism of AlF3 coating on the Li-and Mn-rich cathode materials[J]. Chemistry of Materials, 2014,26(22):6320-6327.
WU Y, MURUGAN A, MANTHIRAM A. Surface modification of high capacity layered Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathodes by AlPO4[J]. Journal of The Electrochemical Society. 2008,155(9):A635-A641.
WANG Q Y, LIU J, MURUGAN A, et al. High capacity double-layer surface modified Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode with improved rate capability[J]. Journal of Materials Chemistry, 2009, 19: 4965-4972.
MIAO X W, NI H, ZHANG H, et al. Li2ZrO3-coated 0.4Li2MnO3·0.6LiNi1/3Co1/3Mn1/3O2 for high performance cathode material in lithium-ion battery[J]. Journal of Power Sources, 2014, 264: 147-154.
LIU X, SU Q, ZHANG C, et al. Enhanced electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2 cathode with an ionic conductive LiVO3 coating layer[J]. ACS Sustainable Chemistry & Engineering, 2015,4(1):255-263.
XIN Y L, QI L Y, ZHANG Y W, et al. Organic solvent-assisted free-standing Li2MnO3·LiNi1/3Co1/3Mn1/3O2 on 3D graphene as a high energy density cathode[J]. Chemical Communications (Cambridge, England), 2015, 51(91): 16381-16384.
YU D Y W, YANAGIDA K, NAKAMURA H. Surface modification of Li-excess Mn-based cathode materials[J]. Journal of the Electrochemical Society, 2010, 157(11): A1177.
GUO H C, WEI Z, JIA K, et al. Abundant nanoscale defects to eliminate voltage decay in Li-rich cathode materials[J]. Energy Storage Materials, 2019, 16: 220-227.
QIU B, ZHANG M H, WU L J, et al. Gas-solid interfacial modification of oxygen activity in layered oxide cathodes for lithium-ion batteries[J]. Nature Communications, 2016, 7: 12108.
ERICKSON E M,SCLAR H,SCHIPPER F, et al. High-temperature treatment of Li-rich cathode materials with ammonia: Improved capacity and mean voltage stability during cycling[J]. Advanced Energy Materials. 2017: doi: 10.1002/aenm.201700708.
CHEN Y F, LIU Y C, ZHANG J C, et al. Constructing O2/O3 homogeneous hybrid stabilizes Li-rich layered cathodes[J]. Energy Storage Materials, 2022, 51: 756-763.
DING X K, LUO D, CUI J X, et al. An ultra-long-life lithium-rich Li1.2Mn0.6Ni0.2O2 cathode by three-in-one surface modification for lithium-ion batteries[J]. Angewandte Chemie International Edition, 2020, 59(20): 7778-7782.
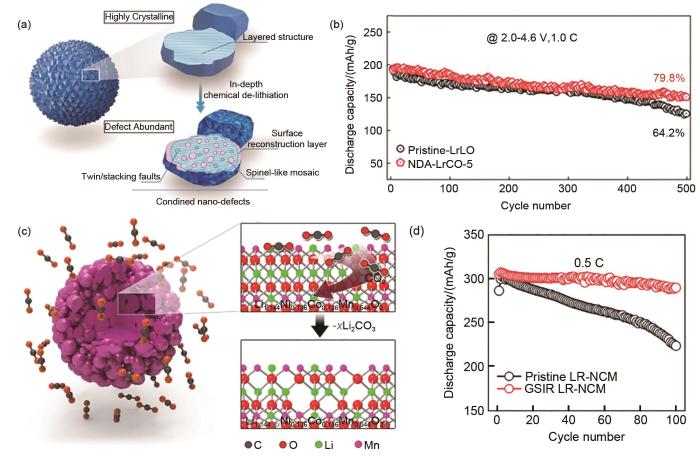
... [7](b);不同方法制备的Li1.2Mn0.6Ni0.2O2 材料的元素分布(c) 及容量循环性能 (d) 和平均电压循环性能[8](e)(a) Active(010)planes and (b) rate capability of Li<sub>1.2</sub>Mn<sub>0.6</sub>Ni<sub>0.2</sub>O<sub>2 </sub>materials<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup>; (c) Element distribution, (d) capacity and (e) average voltage cycle performance of Li<sub>1.2</sub>Mn<sub>0.6</sub>Ni<sub>0.2</sub>O<sub>2 </sub>materials<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>Fig. 53 富锂锰基正极材料的改性研究
... [8](e)(a) Active(010)planes and (b) rate capability of Li<sub>1.2</sub>Mn<sub>0.6</sub>Ni<sub>0.2</sub>O<sub>2 </sub>materials<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup>; (c) Element distribution, (d) capacity and (e) average voltage cycle performance of Li<sub>1.2</sub>Mn<sub>0.6</sub>Ni<sub>0.2</sub>O<sub>2 </sub>materials<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>Fig. 53 富锂锰基正极材料的改性研究
... 由于Na、K元素和Li位于同一主族,早先人们利用Na和K等低价元素对材料进行掺杂改性.Lim等[9]研究了不同Na含量掺杂对Li1.167-x Na x Ni0.18Mn0.548Co0.105O2(0≥x≥0.1)性能的影响,发现当x=0.05时,锂离子扩散系数从1.35×10-9 cm2/s提高到3.34×10-9 cm2/s,容量保持率从83%提高到92%,倍率性能也得到一定的改善.Li等[10]通过采用含有K元素的α-MnO2作为原料,经过原位合成得到K+掺杂的Li1.2Mn0.54Co0.13Ni0.13O2,并对K元素的作用进行了系统性研究,发现半径较大的K+可以降低锂层三空位的形成和锰的迁移,阻碍尖晶石相结构的形成,因此,该材料表现出很好的循环稳定性,循环110周后,容量保持率为85%.进一步研究发现如果Na+或K+在材料中均匀地掺杂,由于位阻效应的存在,使得材料不会从层状结构向尖晶石结构的转变,除非发生明显的晶格畸变.但在长时间循环过程中由于Jahn-Teller畸变会产生Mn3+,Mn3+再发生歧化反应生成Mn2+,Mn2+更倾向于四面体位点形成尖晶石相.因此,Na和K元素掺杂的材料最终还是会形成尖晶石相,只不过相比不掺杂的材料这一过程会被延缓而已. ...
4
... Modification methode and electrochemical performance comparsion of LMR cathode materialsTable 1
... 由于Na、K元素和Li位于同一主族,早先人们利用Na和K等低价元素对材料进行掺杂改性.Lim等[9]研究了不同Na含量掺杂对Li1.167-x Na x Ni0.18Mn0.548Co0.105O2(0≥x≥0.1)性能的影响,发现当x=0.05时,锂离子扩散系数从1.35×10-9 cm2/s提高到3.34×10-9 cm2/s,容量保持率从83%提高到92%,倍率性能也得到一定的改善.Li等[10]通过采用含有K元素的α-MnO2作为原料,经过原位合成得到K+掺杂的Li1.2Mn0.54Co0.13Ni0.13O2,并对K元素的作用进行了系统性研究,发现半径较大的K+可以降低锂层三空位的形成和锰的迁移,阻碍尖晶石相结构的形成,因此,该材料表现出很好的循环稳定性,循环110周后,容量保持率为85%.进一步研究发现如果Na+或K+在材料中均匀地掺杂,由于位阻效应的存在,使得材料不会从层状结构向尖晶石结构的转变,除非发生明显的晶格畸变.但在长时间循环过程中由于Jahn-Teller畸变会产生Mn3+,Mn3+再发生歧化反应生成Mn2+,Mn2+更倾向于四面体位点形成尖晶石相.因此,Na和K元素掺杂的材料最终还是会形成尖晶石相,只不过相比不掺杂的材料这一过程会被延缓而已. ...
... 即使相同的掺杂元素,采用不同的制备方法和工艺也有可能影响元素的掺杂位置和材料的性能.例如,前面提到的Ti4+掺杂在锂位,而日产汽车Yama moto等[16]采用喷雾热解法制备了Ti4+掺杂的Li1.5Ni0.25Mn0.75-x Ti x O2.5,通过TEM和EDX分析证明了Ti均匀地取代了Mn的位置,认为Ti掺杂可以抑制循环过程中Ni和Mn的溶出,因此提高了材料的循环性能. ...
1
... Kim等[17]通过研究F掺杂的0.3Li2MnO3·0.7Li(Mn0.60Ni0.25Co0.15)O1.975F0.025,发现F掺杂能够形成更稳定的SEI层来提高Li+扩散速率,同时减少过渡金属的溶解和降低循环过程中的内阻.Lim等[18]通过研究F掺杂的Li1.2-x Mn0.54Ni0.13Co0.13O2-x F x (x=0.08),认为F取代部分O后可以缓解层状结构向尖晶石结构的转变,降低循环过程中的电压降,提高循环性能.因此,该材料经过100周循环后,容量保持率仍为95%,远高于未掺F材料的容量保持率(62%).此外,F元素和其他金属元素共掺杂也成为了近年来的研究热点,凭借各自的优势互补作用,可以使改性效果更加明显.例如,加州大学伯克利分校的Lee等[19]采用球磨机械化学法合成了共掺杂的岩盐相Li2Mn2/3Nb1/3O2F和Li2Mn1/2Ti1/2O2F两种材料,发现共掺杂可以实现Mn2+/Mn4+的可逆氧化还原反应,因此减少了氧的氧化还原活性,使材料结构稳定.其中,Li2Mn2/3Nb1/3O2F表现出高的容量(>300 mAh/g)和能量密度(约1000 Wh/kg).利用F掺杂到O位可以减小晶格氧的流失,缓解结构的转变,但由于富锂锰基的高容量是由于氧发生了氧化还原反应,因此,F掺杂可能会导致容量有一定降低. ...
1
... Kim等[17]通过研究F掺杂的0.3Li2MnO3·0.7Li(Mn0.60Ni0.25Co0.15)O1.975F0.025,发现F掺杂能够形成更稳定的SEI层来提高Li+扩散速率,同时减少过渡金属的溶解和降低循环过程中的内阻.Lim等[18]通过研究F掺杂的Li1.2-x Mn0.54Ni0.13Co0.13O2-x F x (x=0.08),认为F取代部分O后可以缓解层状结构向尖晶石结构的转变,降低循环过程中的电压降,提高循环性能.因此,该材料经过100周循环后,容量保持率仍为95%,远高于未掺F材料的容量保持率(62%).此外,F元素和其他金属元素共掺杂也成为了近年来的研究热点,凭借各自的优势互补作用,可以使改性效果更加明显.例如,加州大学伯克利分校的Lee等[19]采用球磨机械化学法合成了共掺杂的岩盐相Li2Mn2/3Nb1/3O2F和Li2Mn1/2Ti1/2O2F两种材料,发现共掺杂可以实现Mn2+/Mn4+的可逆氧化还原反应,因此减少了氧的氧化还原活性,使材料结构稳定.其中,Li2Mn2/3Nb1/3O2F表现出高的容量(>300 mAh/g)和能量密度(约1000 Wh/kg).利用F掺杂到O位可以减小晶格氧的流失,缓解结构的转变,但由于富锂锰基的高容量是由于氧发生了氧化还原反应,因此,F掺杂可能会导致容量有一定降低. ...
... Kim等[17]通过研究F掺杂的0.3Li2MnO3·0.7Li(Mn0.60Ni0.25Co0.15)O1.975F0.025,发现F掺杂能够形成更稳定的SEI层来提高Li+扩散速率,同时减少过渡金属的溶解和降低循环过程中的内阻.Lim等[18]通过研究F掺杂的Li1.2-x Mn0.54Ni0.13Co0.13O2-x F x (x=0.08),认为F取代部分O后可以缓解层状结构向尖晶石结构的转变,降低循环过程中的电压降,提高循环性能.因此,该材料经过100周循环后,容量保持率仍为95%,远高于未掺F材料的容量保持率(62%).此外,F元素和其他金属元素共掺杂也成为了近年来的研究热点,凭借各自的优势互补作用,可以使改性效果更加明显.例如,加州大学伯克利分校的Lee等[19]采用球磨机械化学法合成了共掺杂的岩盐相Li2Mn2/3Nb1/3O2F和Li2Mn1/2Ti1/2O2F两种材料,发现共掺杂可以实现Mn2+/Mn4+的可逆氧化还原反应,因此减少了氧的氧化还原活性,使材料结构稳定.其中,Li2Mn2/3Nb1/3O2F表现出高的容量(>300 mAh/g)和能量密度(约1000 Wh/kg).利用F掺杂到O位可以减小晶格氧的流失,缓解结构的转变,但由于富锂锰基的高容量是由于氧发生了氧化还原反应,因此,F掺杂可能会导致容量有一定降低. ...
1
... 在富锂锰基材料表面会涉及很多重要的反应,如电解液的分解、氧气逸出、过渡金属溶解等.近年来,研究发现对材料表面进行包覆处理可以有效地提高LMR材料的表面结构稳定性,降低电压降和提升循环、倍率性能.Wu等[20]发现Al2O3包覆可以明显地降低首次不可逆容量损失和提高放电容量,并且表现出优异的循环稳定性,作者认为Al2O3包覆抑制了材料表面和电解质之间的副反应.Zhao等[21]采用原子层沉积法在Li(Li0.2Mn0.54Ni0.13Co0.13)O2的表面进行了ZrO2和ZnO包覆,并研究了包覆层的厚度和组成对材料性能的影响,发现采用约10 nm LiCoO2和6层ZrO2原子层厚度的双壳层包覆时,材料表现出最高的放电容量(296.4 mAh/g)和优异的循环性能、倍率性能,作者认为该双层功能性包覆的协同效应能够降低循环过程中的电化学极化、结构变化和表面副反应.北京理工大学Wu等[22]通过研究MnO x 包覆的Li(Ni0.2Li0.2Mn0.6)O2,发现10%的MnO x (厚约20 nm)包覆量能够有效地改善材料的电化学性能,作者认为这是由于MnO x 包覆不仅能够保留材料层状结构内部的部分Li空位,本身还可以提供Li+嵌入位点. ...
1
... 在富锂锰基材料表面会涉及很多重要的反应,如电解液的分解、氧气逸出、过渡金属溶解等.近年来,研究发现对材料表面进行包覆处理可以有效地提高LMR材料的表面结构稳定性,降低电压降和提升循环、倍率性能.Wu等[20]发现Al2O3包覆可以明显地降低首次不可逆容量损失和提高放电容量,并且表现出优异的循环稳定性,作者认为Al2O3包覆抑制了材料表面和电解质之间的副反应.Zhao等[21]采用原子层沉积法在Li(Li0.2Mn0.54Ni0.13Co0.13)O2的表面进行了ZrO2和ZnO包覆,并研究了包覆层的厚度和组成对材料性能的影响,发现采用约10 nm LiCoO2和6层ZrO2原子层厚度的双壳层包覆时,材料表现出最高的放电容量(296.4 mAh/g)和优异的循环性能、倍率性能,作者认为该双层功能性包覆的协同效应能够降低循环过程中的电化学极化、结构变化和表面副反应.北京理工大学Wu等[22]通过研究MnO x 包覆的Li(Ni0.2Li0.2Mn0.6)O2,发现10%的MnO x (厚约20 nm)包覆量能够有效地改善材料的电化学性能,作者认为这是由于MnO x 包覆不仅能够保留材料层状结构内部的部分Li空位,本身还可以提供Li+嵌入位点. ...
1
... 在富锂锰基材料表面会涉及很多重要的反应,如电解液的分解、氧气逸出、过渡金属溶解等.近年来,研究发现对材料表面进行包覆处理可以有效地提高LMR材料的表面结构稳定性,降低电压降和提升循环、倍率性能.Wu等[20]发现Al2O3包覆可以明显地降低首次不可逆容量损失和提高放电容量,并且表现出优异的循环稳定性,作者认为Al2O3包覆抑制了材料表面和电解质之间的副反应.Zhao等[21]采用原子层沉积法在Li(Li0.2Mn0.54Ni0.13Co0.13)O2的表面进行了ZrO2和ZnO包覆,并研究了包覆层的厚度和组成对材料性能的影响,发现采用约10 nm LiCoO2和6层ZrO2原子层厚度的双壳层包覆时,材料表现出最高的放电容量(296.4 mAh/g)和优异的循环性能、倍率性能,作者认为该双层功能性包覆的协同效应能够降低循环过程中的电化学极化、结构变化和表面副反应.北京理工大学Wu等[22]通过研究MnO x 包覆的Li(Ni0.2Li0.2Mn0.6)O2,发现10%的MnO x (厚约20 nm)包覆量能够有效地改善材料的电化学性能,作者认为这是由于MnO x 包覆不仅能够保留材料层状结构内部的部分Li空位,本身还可以提供Li+嵌入位点. ...
2
... Modification methode and electrochemical performance comparsion of LMR cathode materialsTable 1
... [33];(d) 多晶和(e) 单晶富锂锰基材料的充放电曲线和产气对比[35](a) STEM image, (b) voltage and (c) capacity cycle performance of O2/O3 type LMR materials<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>; Charge-discharge curve and gas evolution comparison of (d) polycrystal and (e) single crystal LMR materials<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup>Fig. 84 结论与展望
... [35](a) STEM image, (b) voltage and (c) capacity cycle performance of O2/O3 type LMR materials<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>; Charge-discharge curve and gas evolution comparison of (d) polycrystal and (e) single crystal LMR materials<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup>Fig. 84 结论与展望






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}