兼具高能量密度、高功率密度、长循环寿命性能的正极材料是当下电池储能材料研究的重点,也是储能市场的重要需求。富锂锰基正极材料(LRMO)因其极高的放电比容量(≥250 mAh/g)、较高的工作电压(4.2~4.5 V vs. Li/Li+)、低成本且环境友好等优点成为当下最具应用前景的正极材料之一。虽然金属阳离子和阴离子依次或同时进行的氧化还原反应使LRMO材料的容量超过了传统层状氧化物,但首次不可逆容量高、循环和倍率性能较差等一系列的问题阻碍了其工程化应用,这与材料中阴离子氧化还原反应紧密相关。本文首先介绍了LRMO材料的晶体结构,然后基于分子轨道理论,回顾了LRMO材料的能带结构与阴阳离子氧化还原反应的联系,总结了阴离子氧化还原反应对富锂锰基正极材料的影响,包括高容量、不可逆的氧流失、过渡金属离子迁移。同时,分别从过渡金属比例调节、表面修饰、离子掺杂三个方面总结了近些年国内外研究人员针对阴离子氧化还原反应造成的负面影响设计的改性策略。最后展望了LRMO材料理论研究与应用研究的大致方向。
关键词:锂离子电池
;
富锂锰基
;
正极材料
;
阴离子氧化还原
;
改性策略
Abstract
Cathode materials with high energy density, high power density, and long cycle life are the focus of current research on battery energy storage materials and are also in high demand in the energy storage market. Lithium-rich manganese-based oxide (LRMO) cathode materials are some of the most promising cathode materials owing to their high discharge specific capacity (≥250 mAh/g), high operating voltage (4.2~4.5 V vs. Li/Li+), low cost, and environmental friendliness. Although the sequential or simultaneous redox of cations and anions of LRMO materials results in their enhanced capacity compared with other conventional layered oxides, several problems such as high irreversible capacity for the first cycle and poor cycling and rate performance hinder their engineering applications, which are closely related to the anionic redox reactions in the materials. This paper introduces the crystal structure of LRMO materials and then reviews the relationship between the energy band structure of LRMO materials and anionic redox reactions based on molecular orbital theory. In addition, the effects of anionic redox reactions on LRMO cathode materials, including high capacity, irreversible oxygen loss, and transition metal ion migration, are summarized. Moreover, recent modification strategies for mitigating the negative effects of anionic redox reactions are summarized from three perspectives: transition metal ratio adjustment, surface modification, and ion doping. Finally, the paper discusses the future theoretical and application direction of LRMO materials.
Keywords:lithium-ion batteries
;
lithium-rich manganese based
;
cathode material
;
anionic redox
;
modification strategy
ZHOU Junfei. Effect of anionic redox reaction on lithium-rich manganese-based materials and its modification strategy[J]. Energy Storage Science and Technology, 2022, 11(12): 3733-3740
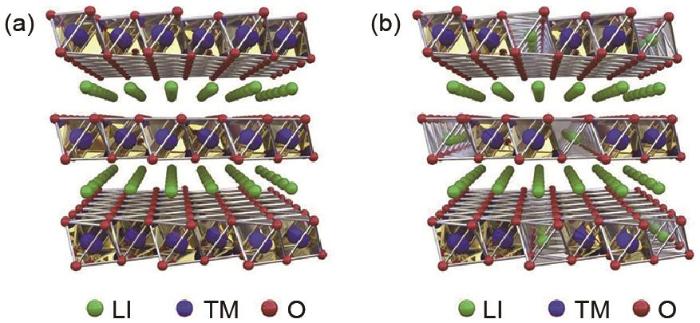
随着电动汽车、5G基站、智能电网等新型储能领域的发展,对锂离子电池的能量密度、功率密度、循环寿命及安全性能都提出了更高的要求。正极材料作为锂离子电池的重要组成部分,开发高能量密度、长循环寿命和高安全性兼具的正极材料成为当下亟待解决的问题之一。目前商用的正极材料,如钴酸锂[1](LiCoO2,140 mAh/g)、磷酸铁锂(LiFePO4,150 mAh/g)、锰酸锂(LiMn2O4,120 mAh/g)和三元材料[2](LiNi x Co y Mn1-x-y O2,220 mAh/g)等尚且无法满足当下需求。1997年,Numata等[3]首次合成了一种锂锰钴氧化物Li(Li x/3Mn2x/3Co1-x )O2(0≤x≤1),可以看成是LiCoO2和Li2MnO3组成的固溶体。在随后的研究中发现,在大于4.5 V的高电压下,锂离子可以从Li2MnO3中脱出 [4],这种特性使得这一类富锂层状氧化物的容量高于传统的层状氧化物,理论容量可高达250 mAh/g成为目前报道中容量最高的正极材料之一[5]。一直以来,富锂正极材料在脱锂过程中过渡金属阳离子被认为会发生氧化还原反应参与电荷补偿来提供容量[6]。但近期研究者发现在Li2MnO3活化时,材料中的阴离子也发生氧化反应参与电荷补偿以提供容量[7]。
WANG J X, LIANG Z, ZHAO Y, et al. Direct conversion of degraded LiCoO2 cathode materials into high-performance LiCoO2: A closed-loop green recycling strategy for spent lithium-ion batteries[J]. Energy Storage Materials, 2022, 45: 768-776.
CHANG Z, QIAO Y, YANG H J, et al. Sustainable lithium-metal battery achieved by a safe electrolyte based on recyclable and low-cost molecular sieve[J]. Angewandte Chemie (International Ed in English), 2021, 60(28): 15572-15581.
NUMATA K, SAKAKI C, YAMANAKA S. Synthesis of solid solutions in a system of LiCoO2-Li2MnO3for cathode materials of secondary lithium batteries[J]. Chemistry Letters, 1997, 26(8): 725-726.
KALYANI P, CHITRA S, MOHAN T, et al. Lithium metal rechargeable cells using Li2MnO3 as the positive electrode[J]. Journal of Power Sources, 1999, 80(1/2): 103-106.
YU H J, SO Y G, KUWABARA A, et al. Crystalline grain interior configuration affects lithium migration kinetics in Li-rich layered oxide[J]. Nano Letters, 2016, 16(5): 2907-2915.
LI X, QIAO Y, GUO S H, et al. Direct visualization of the reversible O2-/O- redox process in Li-rich cathode materials[J]. Advanced Materials, 2018, 30(14): doi: 10.1002/adma.201705197.
ZHENG H F, HAN X, GUO W B, et al. Recent developments and challenges of Li-rich Mn-based cathode materials for high-energy lithium-ion batteries[J]. Materials Today Energy, 2020, 18: doi:10.1016/j.mtener.2020.100518.
YU H J, ISHIKAWA R, SO Y G, et al. Direct atomic-resolution observation of two phases in the Li1.2Mn0.567Ni0.166Co0.067O2 cathode material for lithium-ion batteries[J]. Angewandte Chemie, 2013, 52(23): 5969-5973.
JARVIS K A, DENG Z Q, ALLARD L F, et al. Atomic structure of a lithium-rich layered oxide material for lithium-ion batteries: Evidence of a solid solution[J]. Chemistry of Materials, 2011, 23(16): 3614-3621.
KANG R T, XIAO J, SUN Y N, et al. Research development on the effect of anionic redox reaction on the properties of Li-rich layered materials[J]. Journal of Liaocheng University (Natural Science Edition), 2021, 34(2): 49-58.
SEO D H, LEE J, URBAN A, et al. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials[J]. Nature Chemistry, 2016, 8(7): 692-697.
LI Q Y, DE NING, WONG D, et al. Improving the oxygen redox reversibility of Li-rich battery cathode materials via Coulombic repulsive interactions strategy[J]. Nature Communications, 2022, 13: 1123.
WANG M J. Preparation and modification of lithium rich manganese based cathode materials for lithium ion battery[D]. Harbin: Harbin Institute of Technology, 2019.
CHEN Q, PEI Y, CHEN H W, et al. Highly reversible oxygen redox in layered compounds enabled by surface polyanions[J]. Nature Communications, 2020, 11: 3411.
HOUSE R A, REES G J, PÉREZ-OSORIO M A, et al. First-cycle voltage hysteresis in Li-rich 3d cathodes associated with molecular O2 trapped in the bulk[J]. Nature Energy, 2020, 5(10): 777-785.
GENT W E, LIM K, LIANG Y F, et al. Coupling between oxygen redox and cation migration explains unusual electrochemistry in lithium-rich layered oxides[J]. Nature Communications, 2017, 8: 2091.
XU B, FELL C R, CHI M F, et al. Identifying surface structural changes in layered Li-excess nickel manganese oxides in high voltage lithium ion batteries: A joint experimental and theoretical study[J]. Energy & Environmental Science, 2011, 4(6): 2223.
EUM D, KIM B, KIM S J, et al. Voltage decay and redox asymmetry mitigation by reversible cation migration in lithium-rich layered oxide electrodes[J]. Nature Materials, 2020, 19(4): 419-427.
MOHANTY D, HUQ A, PAYZANT E A, et al. Neutron diffraction and magnetic susceptibility studies on a high-voltage Li1.2Mn0.55Ni0.15Co0.10O2 lithium ion battery cathode: Insight into the crystal structure[J]. Chemistry of Materials, 2013, 25(20): 4064-4070.
CUI S L, WANG Y Y, LIU S, et al. Evolution mechanism of phase transformation of Li-rich cathode materials in cycling[J]. Electrochimica Acta, 2019, 328: doi: 10.1016/j.electacta.2019.135109.
XIE Y, YIN J, CHEN X, et al. Synergistic effect of Mn3+ formation-migration and oxygen loss on the near surface and bulk structural changes in single crystalline lithium-rich oxides[J]. ACS Applied Materials & Interfaces, 2021, 13(3): 3891-3898.
VIVEKANANTHA M, SUNDHAR ARUL SARAVANAN R, KUMAR NAYAK P, et al. Synergistic-effect of high Ni content and Na dopant towards developing a highly stable Li-Rich cathode in Li-ion batteries[J]. Chemical Engineering Journal, 2022, 444: doi: 10.1016/j.cej.2022.136503.
ALI S E, OLSZEWSKI W, SORRENTINO A, et al. Local interactions governing the performances of lithium- and manganese-rich cathodes[J]. The Journal of Physical Chemistry Letters, 2021, 12(4): 1195-1201.
SHEN S Y, HONG Y H, ZHU F C, et al. Tuning electrochemical properties of Li-rich layered oxide cathode by adjusting Co/Ni ratio and mechanism investigation using in situ XRD and OEMS[J]. ACS Applied Materials & Interfaces, 2018 10(15):12666-12677.
LI Q Y, NING D, ZHOU D, et al. The effect of oxygen vacancy and spinel phase integration on both anionic and cationic redox in Li-rich cathode materials[J]. Journal of Materials Chemistry A, 2020, 8(16): 7733-7745.
PEI Y, CHEN Q, WANG M Y, et al. Reviving reversible anion redox in 3d-transition-metal Li rich oxides by introducing surface defects[J]. Nano Energy, 2020, 71: doi: 10.1016/j.nanoen.2020.104644.
LIU J D, WU Z H, YU M, et al. Building homogenous Li2TiO3 coating layer on primary particles to stabilize Li-rich Mn-based cathode materials[J]. Small, 2022, 18(10): doi: 10.1002/smll.202106337.
YU H, GAO Y, LIANG X H. Slightly fluorination of Al2O3 ALD coating on Li1.2Mn0.54Co0.13Ni0.13O2 electrodes: Interface reaction to create stable solid permeable interphase layer[J]. Journal of the Electrochemical Society, 2019, 166(10): A2021-A2027.
RASTGOO-DEYLAMI M, JAVANBAKHT M, OMIDVAR H. Enhanced performance of layered Li1.2Mn0.54Ni0.13Co0.13O2 cathode material in Li-ion batteries using nanoscale surface coating with fluorine-doped anatase TiO2[J]. Solid State Ionics, 2019, 331: 74-88.
ZHOU Z W, LUO Z Y, HE Z J, et al. Suppress voltage decay of lithium-rich materials by coating layers with different crystalline states[J]. Journal of Energy Chemistry, 2021, 60: 591-598.
DING X, LI Y X, CHEN F, et al. In situ formation of LiF decoration on a Li-rich material for long-cycle life and superb low-temperature performance[J]. Journal of Materials Chemistry A, 2019, 7(18): 11513-11519.
NIU B B, LI J L, LIU Y Y, et al. Re-understanding the function mechanism of surface coating: Modified Li-rich layered Li1.2Mn0.54Ni0.13Co0.13O2 cathodes with YF3 for high performance lithium-ions batteries[J]. Ceramics International, 2019, 45(9): 12484-12494.
SU Y F, YUAN F Y, CHEN L, et al. Enhanced high-temperature performance of Li-rich layered oxide via surface heterophase coating[J]. Journal of Energy Chemistry, 2020, 51: 39-47.
NIE X K, HOU G M, XU Z, et al. Lewis acidity organoboron-modified Li-rich cathode materials for high-performance lithium-ion batteries[J]. Advanced Materials Interfaces, 2021, 8(9): doi: 10.1002/admi.202002113.
YIN C, WEN X H, WAN L Y, et al. Surface reinforcement doping to suppress oxygen release of Li-rich layered oxides[J]. Journal of Power Sources, 2021, 503: doi: 10.1016/j.jpowsour.2021.230048.
MENG J X, XU L S, MA Q X, et al. Modulating crystal and interfacial properties by W-gradient doping for highly stable and long life Li-rich layered cathodes[J]. Advanced Functional Materials, 2022, 32(19): doi: 10.1002/adfm.202113013.
HU K H, REN L, FAN W F, et al. Tuning redox activity through delithiation induced protective layer and Fe-O coordination for Li-rich cathode with improved voltage and cycle performance[J]. Journal of Energy Chemistry, 2022, 71: 266-276.
LI S Y, FU X L, LIANG Y W, et al. Enhanced structural stability of boron-doped Layered@Spinel@Carbon heterostructured lithium-rich manganese-based cathode materials[J]. ACS Sustainable Chemistry & Engineering, 2020, 8(25): 9311-9324.
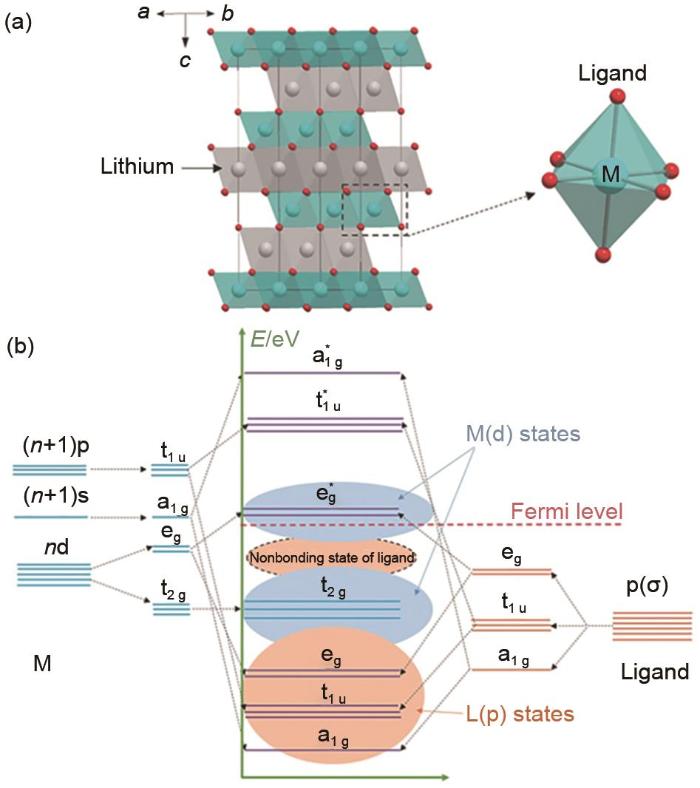
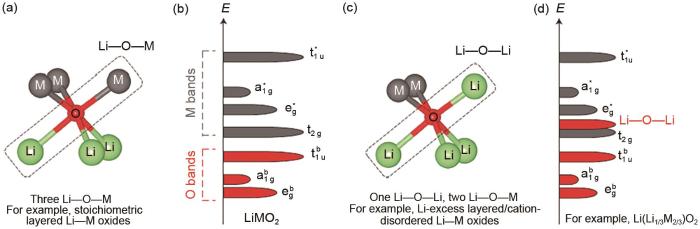
... [11]The energy band structure of the transition metal ion (M) and the ligand (L)<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 23 阴离子氧化还原反应对富锂锰基材料的影响<strong>3.1</strong> 高容量
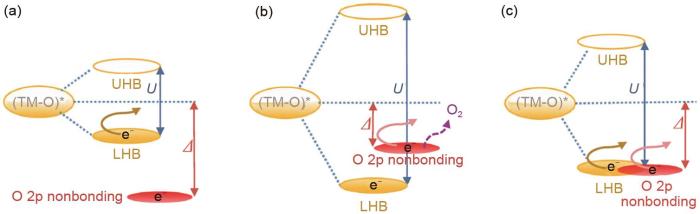
... [14]<strong>Qualitative analysis of the relationship between the d-d Coulombic interaction term <i>U</i> and the charge transfer term</strong><i>Δ</i><sup>[<xref ref-type="bibr" rid="R14">14</xref>]</sup>Fig. 4<strong>3.2</strong> 不可逆的氧流失
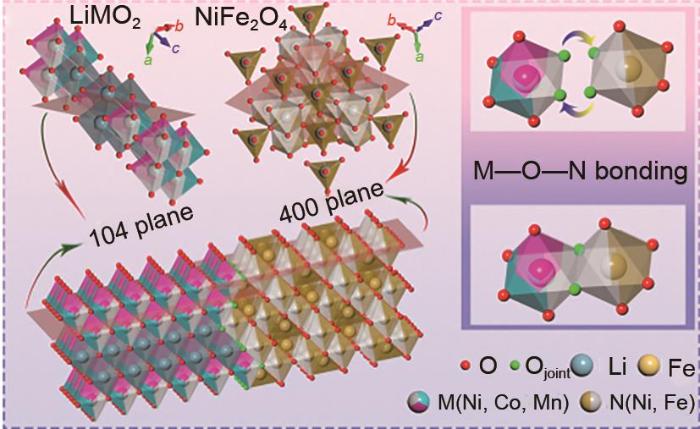
... [39]Schematic diagram of the heterostructure O-shared bonding of LNCM (104 plane) and NFO (400 plane)<sup>[<xref ref-type="bibr" rid="R39">39</xref>]</sup>Fig. 5<strong>4.3</strong> 离子掺杂




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}