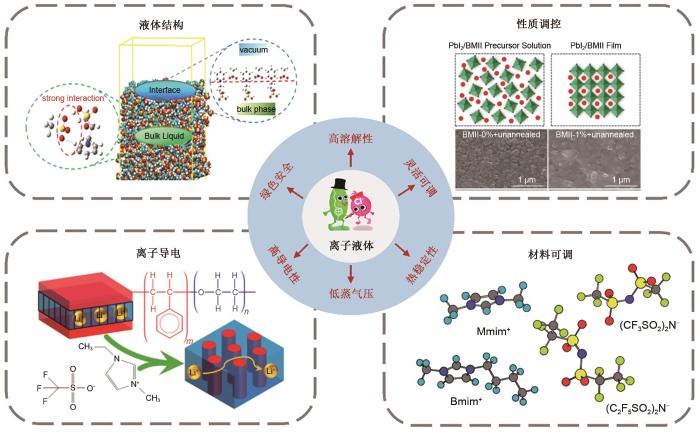
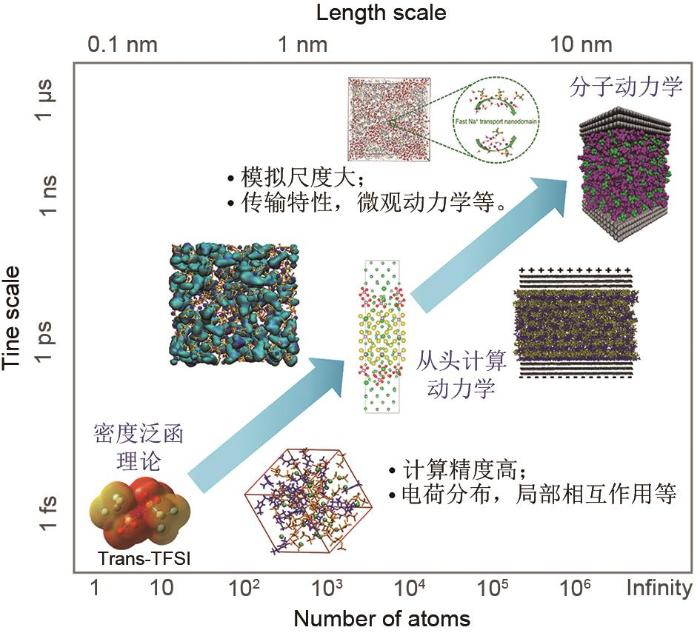
As an important part of electrochemical energy storage system, electrolyte is one of the key factors to determine the battery capacity, support the energy storage and cycle stability of supercapacitor. As a new kind of soft functional materials, ionic liquids are widely used in electrochemical energy storage components, such as lithium batteries and supercapacitors, and gradually become one of the best substitutes for traditional organic electrolytes because of their high conductivity, wide electrochemical window, good thermal stability and no significant vapor pressure. At present, the design and research of ionic liquid electrolytes mostly use experimental test method, which has large search range, high cost, and it is difficult to accurately obtain a deep understanding of its dynamic structure, formation mechanism and action mechanism at the nano and micro level. Therefore, this review aims to summarize the related progress of ionic liquid electrolyte in simulation calculation. Firstly, according to different simulation scales, three simulation methods for ionic liquid electrolytes are introduced, and their advantages and disadvantages are discussed. Secondly, according to the different components of ionic liquid in electrolytes, the simulation research status of ionic liquid in battery and supercapacitor are reviewed respectively. Finally, the future challenges and development direction of ionic liquid electrolytes are discussed, in order to provide a new research idea for the simulation of electrolytes.
Fig.4
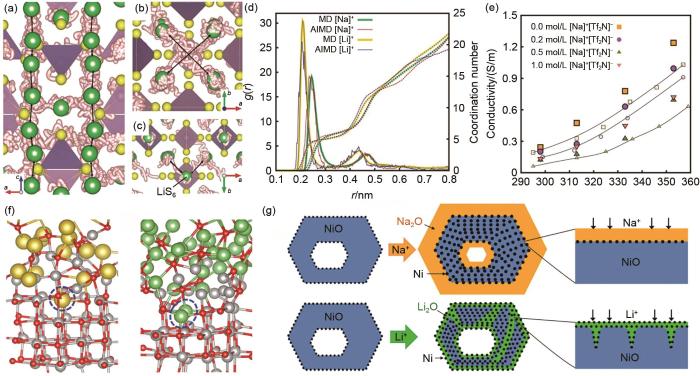
Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD[25]; (d) radial distribution function between Na+/Li+ and [Tf2N]-, from AIMD and MD[26]; (e) conductivity of ionic liquid electrolyte containing Na+[Tf2N]- and Li+[Tf2N]-[26]; (f) snapshots of sodiated and lithiated NiO surfaces[27]; (g) schematic cartoons showing different reaction modes between sodiation and lithiation[27]
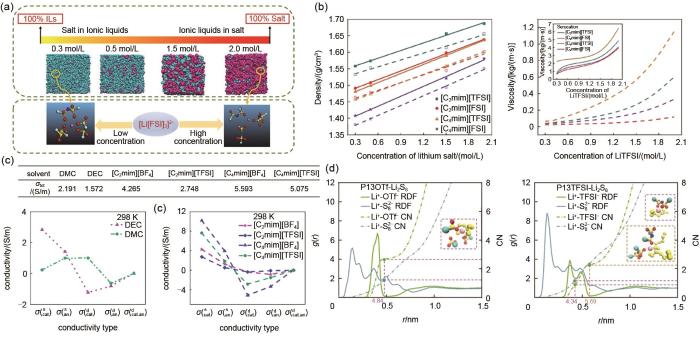
MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质。例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)]。研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合。同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移。随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构。如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低。此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制。因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为。图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度。并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解。同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构。相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比。
图5
MD在电解液中的应用:(a) MD捕获得到的不同锂盐浓度下的模型图[31];(b) 离子液体电解液([C n mim][TFSI]和[C n mim][FSI], n = 2, 4)的密度和黏度与LiTFSI浓度的关系[31];(c) 有机溶剂电解液和离子液体电解液的电导率曲线[32];(d) [P13][OTf]-Li2S8 和[P13][TFSI]-Li2S8 中Li--S82- 和Li+-anions的质心RDF及配位数曲线[33]
Fig. 5
Application of MD in electrolyte (a) Snapshots of different lithium concentrations from MD[31]; (b) Density and viscosity vs. concentration of LiTFSI for all ionic liquid electrolytes ([C n mim][TFSI] and [C n mim][FSI], n = 2, 4)[31]; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte[32]; (d) Center-of-mass RDFs and coordination number curves of Li+-S82- and Li+-anions for [P13][OTf]-Li2S8and [P13][TFSI]-Li2S8 systems[33]
Fig. 6
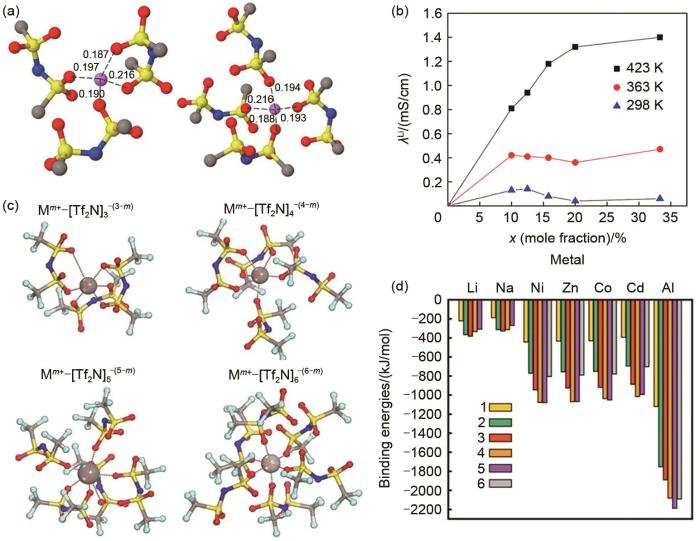
(a) two Li+ cation solvation structures diagrammatic drawing and (b) Li+ contribution to ionic conductivity of ionic liquid electrolytes ([Pyr13][TFSI]/LiTFSI)[37]; (c) representative ion clusters from small MD simulations and (d) binding energies of ion clusters[38]
Fig. 7
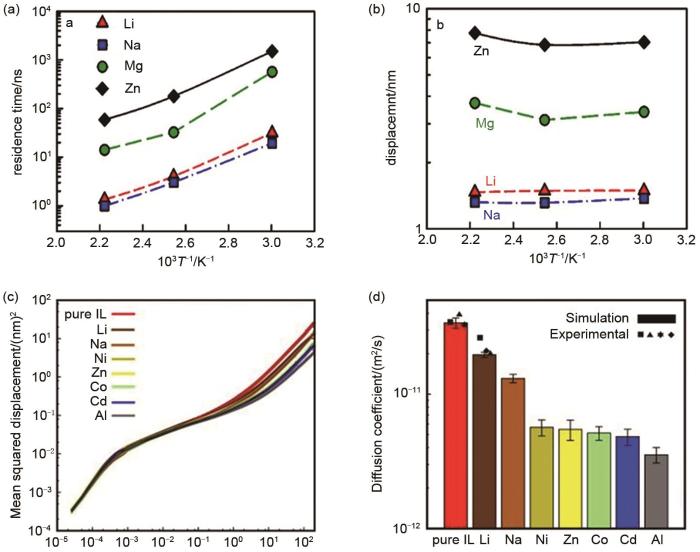
(a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr14][TFSI]/MeTFSI (Me= Li+, Na+, Mg+, Zn2+)[40]; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr14] of [Pyr14][TFSI] / MeTFSI (Me= Li+, Ni2+)[41]
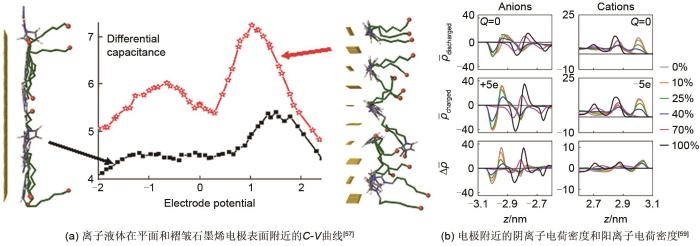
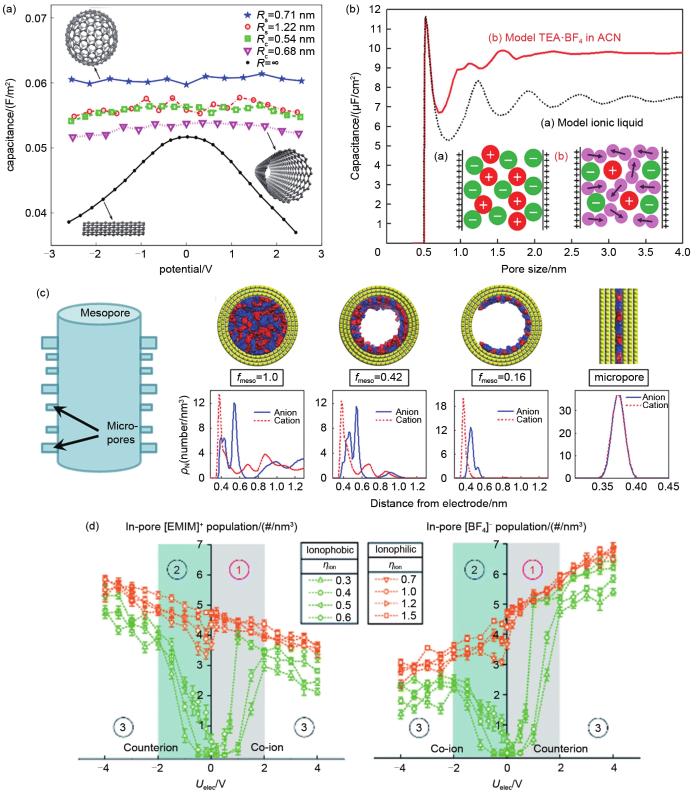
石墨烯材料具有高效吸附电解液等特性,离子液体在电极带电表面吸附的行为及离子化学结构和电双层结构间的函数关系至关重要,并且其结构简单,是一种典型的平板电极电容器。如图8(a)所示,Bedrov等[57]证明离子液体([C n mim][TFSI], n=2, 4, 6, 8)在平面和褶皱面石墨烯电极表面附近的C-V曲线均呈驼峰状。差分电容和离子液体电解液结构间的潜在机理值得进一步研究,离子液体类型可能是影响因素之一。随后,Bedrov课题组[58]比较了[Bmim][BF4]和[Bmim][PF6]这两种离子液体,发现不对称性会增加电容极大值和极小值间的差距。为了更准确地描述石墨烯基电极的空间结构和化学结构,Jung等[59]在2014年研究了平行平板电容器中石墨烯的氧化程度对电容的影响。通过理解离子液体电荷重组对电极充电的响应以研究电极的羟基氧化如何影响离子液体对电极电荷的筛选,如图8(b)所示。随着氧化率的增加,离子密度重组随电极氧化而减弱。氧化率为70%时,羟基基团中的氧原子产生的空间位阻使双电层间隙开始增大。因此,离子液体电荷重组能力的降低和双层膜间隙的扩大导致电容与石墨烯的氧化呈负相关。通过直接考虑充电过程可对离子层结构进行分析,Jung课题组[60]研究了氰基离子液体[Emim][SCN]和石墨烯双电层电容器的充电机理,不仅证明电极电荷与相邻离子之间的相关性是非常重要的,而且发现无法通过单一的指数函数拟合电极电荷密度定量分析充电动力学,这是由于超级电容器在充电过程中的电容受到离子液体动力学不均一性的影响变化不恒定所导致的,离子对电容器的电性能起着重要作用。二维层状过渡金属碳化物和氮化物(MXenes)由于具有柔性特征可以进行自由收缩,保证了离子在层内的快速运输,这种动态电荷储存机制对电化学性质的影响十分重要,是近几年研究的重点问题。Simon等[61]在恒充放电条件下采用电极自由移动的MD模拟方法研究了Ti3C2T x MXene电极和[Emim][TFSI]离子液体组成的电容器,对正极和负极的充电/放电机制进行描述拟合实验测量的电极体积变化。正极主要通过离子交换过程实现电荷存储,而负极主要通过反离子插层过程实现。为了了解离子的微观动力学和电化学性能,Mamontov等[62]通过MD模拟研究水的吸附对离子迁移率的影响,尽管MXene/[Emim][TFSI]吸收了大量的水,但大多数水分子都附着在MXene表面,仅有较少的水分子进入内部离子液体中,因此湿度对离子扩散率的影响较小。
Fig. 9
Simulation of ionic liquid electrolytes in porous electrode capacitor: (a) influence of electrode curvature on differential capacitance[64]; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte[65]; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI][66]; (d) in-pore cation and anion population at electrodes[67]
LOURENÇO T C, ZHANG Y, COSTA L T, et al. A molecular dynamics study of lithium-containing aprotic heterocyclic ionic liquid electrolytes[J]. The Journal of Chemical Physics, 2018, 148(19): doi: 10.1063/1.5016276.
DONG K, ZHANG S J, WANG J J. Understanding the hydrogen bonds in ionic liquids and their roles in properties and reactions[J]. Chemical Communications, 2016, 52(41): 6744-6764.
TSUZUKI S, SHINODA W, SAITO H, et al. Molecular dynamics simulations of ionic liquids: Cation and anion dependence of self-diffusion coefficients of ions[J]. The Journal of Physical Chemistry B, 2009, 113(31): 10641-10649.
NIU T T, CHAO L F, GAO W Y, et al. Ionic liquids-enabled efficient and stable perovskite photovoltaics: Progress and challenges[J]. ACS Energy Letters, 2021, 6(4): 1453-1479.
METWALLI E, KAEPPEL M V, SCHAPER S J, et al. Conductivity and morphology correlations of ionic-liquid/lithium-salt/block copolymer nanostructured hybrid electrolytes[J]. ACS Applied Energy Materials, 2018, 1(2): 666-675.
SHAN W D, YANG Q W, SU B G, et al. Proton microenvironment and interfacial structure of sulfonic-acid-functionalized ionic liquids[J]. The Journal of Physical Chemistry C, 2015, 119(35): 20379-20388.
HAKIM L, ISHII Y, MATSUMOTO K, et al. Transport properties of ionic liquid and sodium salt mixtures for sodium-ion battery electrolytes from molecular dynamics simulation with a self-consistent atomic charge determination[J]. The Journal of Physical Chemistry B, 2020, 124(33): 7291-7305.
CHEN F F, HOWLETT P, FORSYTH M. Na-ion solvation and high transference number in superconcentrated ionic liquid electrolytes: A theoretical approach[J]. The Journal of Physical Chemistry C, 2018, 122(1): 105-114.
RAJPUT N N, MONK J, HUNG F R. Structure and dynamics of an ionic liquid confined inside a charged slit graphitic nanopore[J]. The Journal of Physical Chemistry C, 2012, 116(27): 14504-14513.
SINGH R, MONK J, HUNG F R. Heterogeneity in the dynamics of the ionic liquid[BMIM+][PF6–] confined in a slit nanopore[J]. The Journal of Physical Chemistry C, 2011, 115(33): 16544-16554.
CHENG T, MERINOV B V, MOROZOV S, et al. Quantum mechanics reactive dynamics study of solid Li-electrode/Li6PS5Cl-electrolyte interface[J]. ACS Energy Letters, 2017, 2(6): 1454-1459.
JIANG L W, LIU L L, YUE J M, et al. High-voltage aqueous Na-ion battery enabled by inert-cation-assisted water-in-salt electrolyte[J]. Advanced Materials, 2020, 32(2): doi: 10.1002/adma.201904427.
DUBNIKOVA F, ZEIRI Y. Structure, energies, and vibrational frequencies of solvated Li+ in ionic liquids: Role of cation type[J]. The Journal of Physical Chemistry A, 2016, 120(19): 3079-3087.
SODEYAMA K, YAMADA Y, AIKAWA K, et al. Sacrificial anion reduction mechanism for electrochemical stability improvement in highly concentrated Li-salt electrolyte[J]. The Journal of Physical Chemistry C, 2014, 118(26): 14091-14097.
HARPER A F, EVANS M L, DARBY J P, et al. Ab initio structure prediction methods for battery materials: A review of recent computational efforts to predict the atomic level structure and bonding in materials for rechargeable batteries[J]. Johnson Matthey Technology Review, 2020, 64(2): 103-118.
ONG S P, ANDREUSSI O, WU Y B, et al. Electrochemical windows of room-temperature ionic liquids from molecular dynamics and density functional theory calculations[J]. Chemistry of Materials, 2011, 23(11): 2979-2986.
MONTI D, JÓNSSON E, PALACÍN M R, et al. Ionic liquid based electrolytes for sodium-ion batteries: Na+ solvation and ionic conductivity[J]. Journal of Power Sources, 2014, 245: 630-636.
CLARKE-HANNAFORD J, BREEDON M, RÜTHER T, et al. Spectroscopic and computational study of boronium ionic liquids and electrolytes[J]. Chemistry, 2021, 27(50): 12826-12834.
HE X F, ZHU Y Z, EPSTEIN A, et al. Statistical variances of diffusional properties from ab initio molecular dynamics simulations[J]. npj Computational Materials, 2018, 4:18.
MO Y F, ONG S P, CEDER G. First principles study of the Li10GeP2S12 lithium super ionic conductor material[J]. Chemistry of Materials, 2012, 24(1): 15-17.
VICENT-LUNA J M, ORTIZ-ROLDAN J M, HAMAD S, et al. Quantum and classical molecular dynamics of ionic liquid electrolytes for Na/Li-based batteries: Molecular origins of the conductivity behavior[J]. ChemPhysChem, 2016, 17(16): 2473-2481.
HE K, LIN F, ZHU Y Z, et al. Sodiation kinetics of metal oxide conversion electrodes: A comparative study with lithiation[J]. Nano Letters, 2015, 15(9): 5755-5763.
THOMAS M, BREHM M, HOLLÓCZKI O, et al. Simulating the vibrational spectra of ionic liquid systems: 1-ethyl-3-methylimidazolium acetate and its mixtures[J]. The Journal of Chemical Physics, 2014, 141(2): doi: 10.1063/1.4887082.
BEDROV D, PIQUEMAL J P, BORODIN O, et al. Molecular dynamics simulations of ionic liquids and electrolytes using polarizable force fields[J]. Chemical Reviews, 2019, 119(13): 7940-7995.
TONG J H, WU S L, SOLMS N, et al. The effect of concentration of lithium salt on the structural and transport properties of ionic liquid-based electrolytes[J]. Frontiers in Chemistry, 2020, 7: doi: 10.3389/fchem.2019.00945.
TONG J H, XIAO X Q, LIANG X D, et al. Insights into the solvation and dynamic behaviors of a lithium salt in organic- and ionic liquid-based electrolytes[J]. Physical Chemistry Chemical Physics: PCCP, 2019, 21(35): 19216-19225.
HU T Y, WANG Y L, HUO F, et al. Understanding structural and transport properties of dissolved Li2S8 in ionic liquid electrolytes through molecular dynamics simulations[J]. Chemphyschem, 2021, 22(4): 419-429.
HU T Y, WANG Y L, HUO F, et al. Molecular dynamics simulations of short-chain lithium polysulfides clustering in ionic liquids[J]. The Chinese Journal of Process Engineering, 2021, 21(7): 847-856.
RAY P, BALDUCCI A, KIRCHNER B. Molecular dynamics simulations of lithium-doped ionic-liquid electrolytes[J]. The Journal of Physical Chemistry B, 2018, 122(46): 10535-10547.
BORODIN O, SMITH G D, HENDERSON W. Li+ cation environment, transport, and mechanical properties of the LiTFSI doped N-methyl-N-alkylpyrrolidinium+TFSI- ionic liquids[J]. The Journal of Physical Chemistry B, 2006, 110(34): 16879-16886.
LI Z, SMITH G D, BEDROV D. Li+ solvation and transport properties in ionic liquid/lithium salt mixtures: A molecular dynamics simulation study[J]. The Journal of Physical Chemistry B, 2012, 116(42): 12801-12809.
VICENT-LUNA J M, AZACETA E, HAMAD S, et al. Molecular dynamics analysis of charge transport in ionic-liquid electrolytes containing added salt with mono, di, and trivalent metal cations[J]. Chemphyschem, 2018, 19(13): 1665-1673.
LESCH V, LI Z, BEDROV D, et al. The influence of cations on lithium ion coordination and transport in ionic liquid electrolytes: A MD simulation study[J]. Physical Chemistry Chemical Physics: PCCP, 2016, 18(1): 382-392.
LIU H J, MAGINN E. Effect of ion structure on conductivity in lithium-doped ionic liquid electrolytes: A molecular dynamics study[J]. The Journal of Chemical Physics, 2013, 139(11): doi: 10.1063/1.4821155.
BORODIN O, GIFFIN G A, MORETTI A, et al. Insights into the structure and transport of the lithium, sodium, magnesium, and zinc bis(trifluoromethansulfonyl) imide salts in ionic liquids[J]. The Journal of Physical Chemistry C, 2018, 122(35): 20108-20121.
KUBISIAK P, WRÓBEL P, EILMES A. Molecular dynamics investigation of correlations in ion transport in MeTFSI/EMIM-TFSI (Me = Li, Na) electrolytes[J]. The Journal of Physical Chemistry B, 2020, 124(2): 413-421.
TU K M, ISHIZUKA R, MATUBAYASI N. Spatial-decomposition analysis of electrical conductivity in concentrated electrolyte solution[J]. The Journal of Chemical Physics, 2014, 141(4): doi: 10.1002/tcr.201800116.
SCHRÖDER C, HABERLER M, STEINHAUSER O. On the computation and contribution of conductivity in molecular ionic liquids[J]. The Journal of Chemical Physics, 2008, 128(13): doi: 10.1063/1.2868752.
WOHDE F, BALABAJEW M, ROLING B. Li+Transference numbers in liquid electrolytes obtained by very-low-frequency impedance spectroscopy at variable electrode distances[J]. Journal of the Electrochemical Society, 2016, 163(5): A714-A721.
SHIGENOBU K, DOKKO K, WATANABE M, et al. Solvent effects on Li ion transference number and dynamic ion correlations in glyme- and sulfolane-based molten Li salt solvates[J]. Physical Chemistry Chemical Physics, 2020, 22(27): 15214-15221.
UENO K, YOSHIDA K, TSUCHIYA M, et al. Glyme-lithium salt equimolar molten mixtures: Concentrated solutions or solvate ionic liquids? [J]. The Journal of Physical Chemistry B, 2012, 116(36): 11323-11331.
MANDAI T, YOSHIDA K, TSUZUKI S, et al. Effect of ionic size on solvate stability of glyme-based solvate ionic liquids[J]. The Journal of Physical Chemistry B, 2015, 119(4): 1523-1534.
TERADA S, MANDAI T, SUZUKI S, et al. Thermal and electrochemical stability of tetraglyme-magnesium bis (trifluoromethanesulfonyl) amide complex: Electric field effect of divalent cation on solvate stability[J]. The Journal of Physical Chemistry C, 2016, 120(3): 1353-1365.
SUO L M, BORODIN O, GAO T, et al. "Water-in-salt" electrolyte enables high-voltage aqueous lithium-ion chemistries[J]. Science, 2015, 350(6263): 938-943.
KO S, YAMADA Y, MIYAZAKI K, et al. Lithium-salt monohydrate melt: A stable electrolyte for aqueous lithium-ion batteries[J]. Electrochemistry Communications, 2019, 104: doi: 10.1016/j.elecom.2019.106488.
CHEN L, ZHANG J X, LI Q, et al. A 63 m superconcentrated aqueous electrolyte for high-energy Li-ion batteries[J]. ACS Energy Letters, 2020, 5(3): 968-974.
DOU Q Y, WANG Y, WANG A P, et al. "Water in salt/ionic liquid" electrolyte for 2.8 V aqueous lithium-ion capacitor[J]. Science Bulletin, 2020, 65(21): 1812-1822.
BECKER M, RENTSCH D, REBER D, et al. The hydrotropic effect of ionic liquids in water-in-salt electrolytes[J]. Angewandte Chemie International Edition, 2021, 60(25): 14100-14108.
JORALEECHANCHAI N, DUANGDANGCHOTE S, SAWANGPHRUK M. Insight into the effect of ionic liquid-based additives at the solid electrolyte interphase for lithium metal batteries[J]. Journal of the Electrochemical Society, 2021, 168(4): doi: 10.1149/1945-7111/abf7e3.
VATAMANU J, BORODIN O, BEDROV D, et al. Molecular dynamics simulation study of the interfacial structure and differential capacitance of alkylimidazolium bis(trifluoromethanesulfonyl) imide [Cnmim][TFSI] ionic liquids at graphite electrodes[J]. The Journal of Physical Chemistry C, 2012, 116(14): 7940-7951.
HU Z Z, VATAMANU J, BORODIN O, et al. A molecular dynamics simulation study of the electric double layer and capacitance of [BMIM][PF6] and [BMIM][BF4] room temperature ionic liquids near charged surfaces[J]. Physical Chemistry Chemical Physics: PCCP, 2013, 15(34): 14234-14247.
DEYOUNG A D, PARK S W, DHUMAL N R, et al. Graphene oxide supercapacitors: A computer simulation study[J]. The Journal of Physical Chemistry C, 2014, 118(32): 18472-18480.
NOH C, JUNG Y. Understanding the charging dynamics of an ionic liquid electric double layer capacitor via molecular dynamics simulations[J]. Physical Chemistry Chemical Physics: PCCP, 2019, 21(13): 6790-6800.
XU K, LIN Z F, MERLET C, et al. Tracking ionic rearrangements and interpreting dynamic volumetric changes in two-dimensional metal carbide supercapacitors: A molecular dynamics simulation study[J]. ChemSusChem, 2018, 11(12): 1892-1899.
OSTI N C, THOMPSON M W, VAN AKEN K L, et al. Humidity exposure enhances microscopic mobility in a room-temperature ionic liquid in MXene[J]. The Journal of Physical Chemistry C, 2018, 122(48): 27561-27566.
AKEN K L, MCDONOUGH J K, LI S, et al. Effect of cation on diffusion coefficient of ionic liquids at onion-like carbon electrodes[J]. Journal of Physics Condensed Matter, 2014, 26(28): doi: 10.1088/0953-8984/26/28/284104.
FENG G, LI S, PRESSER V, et al. Molecular insights into carbon supercapacitors based on room-temperature ionic liquids[J]. The Journal of Physical Chemistry Letters, 2013, 4(19): 3367-3376.
JIANG D E, WU J Z. Microscopic insights into the electrochemical behavior of nonaqueous electrolytes in electric double-layer capacitors[J]. The Journal of Physical Chemistry Letters, 2013, 4(8): 1260-1267.
BAÑUELOS J L, FENG G, FULVIO P F, et al. Densification of ionic liquid molecules within a hierarchical nanoporous carbon structure revealed by small-angle scattering and molecular dynamics simulation[J]. Chemistry of Materials, 2014, 26(2): 1144-1153.
GAN Z D, WANG Y L, WANG M, et al. Ionophobic nanopores enhancing the capacitance and charging dynamics in supercapacitors with ionic liquids[J]. Journal of Materials Chemistry A, 2021, 9(29): 15985-15992.
... [25];(d) 由AIMD和MD得到的Na+/Li+ 和[TFSI]- 间RDF及配位数曲线[26];(e) Na+[TFSI]- 和Li+[TFSI]- 电解液的电导率曲线[26];(f) NiO表面钠化和锂化模型[27];(g) 钠化和锂化的反应机理[27]Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>; (d) radial distribution function between Na<sup>+</sup>/Li<sup>+</sup> and [Tf<sub>2</sub>N]<sup>-</sup>, from AIMD and MD<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>; (e) conductivity of ionic liquid electrolyte containing Na<sup>+</sup>[Tf<sub>2</sub>N]<sup>-</sup> and Li<sup>+</sup>[Tf<sub>2</sub>N]<sup>-[26]</sup>; (f) snapshots of sodiated and lithiated NiO surfaces<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>; (g) schematic cartoons showing different reaction modes between sodiation and lithiation<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>Fig.4
... [25]; (d) radial distribution function between Na+/Li+ and [Tf2N]-, from AIMD and MD[26]; (e) conductivity of ionic liquid electrolyte containing Na+[Tf2N]- and Li+[Tf2N]-[26]; (f) snapshots of sodiated and lithiated NiO surfaces[27]; (g) schematic cartoons showing different reaction modes between sodiation and lithiation[27]Fig.4
... [26];(e) Na+[TFSI]- 和Li+[TFSI]- 电解液的电导率曲线[26];(f) NiO表面钠化和锂化模型[27];(g) 钠化和锂化的反应机理[27]Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>; (d) radial distribution function between Na<sup>+</sup>/Li<sup>+</sup> and [Tf<sub>2</sub>N]<sup>-</sup>, from AIMD and MD<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>; (e) conductivity of ionic liquid electrolyte containing Na<sup>+</sup>[Tf<sub>2</sub>N]<sup>-</sup> and Li<sup>+</sup>[Tf<sub>2</sub>N]<sup>-[26]</sup>; (f) snapshots of sodiated and lithiated NiO surfaces<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>; (g) schematic cartoons showing different reaction modes between sodiation and lithiation<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>Fig.4
... [26];(f) NiO表面钠化和锂化模型[27];(g) 钠化和锂化的反应机理[27]Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>; (d) radial distribution function between Na<sup>+</sup>/Li<sup>+</sup> and [Tf<sub>2</sub>N]<sup>-</sup>, from AIMD and MD<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>; (e) conductivity of ionic liquid electrolyte containing Na<sup>+</sup>[Tf<sub>2</sub>N]<sup>-</sup> and Li<sup>+</sup>[Tf<sub>2</sub>N]<sup>-[26]</sup>; (f) snapshots of sodiated and lithiated NiO surfaces<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>; (g) schematic cartoons showing different reaction modes between sodiation and lithiation<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>Fig.4
... [26]; (e) conductivity of ionic liquid electrolyte containing Na+[Tf2N]- and Li+[Tf2N]-[26]; (f) snapshots of sodiated and lithiated NiO surfaces[27]; (g) schematic cartoons showing different reaction modes between sodiation and lithiation[27]Fig.4
... [27];(g) 钠化和锂化的反应机理[27]Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>; (d) radial distribution function between Na<sup>+</sup>/Li<sup>+</sup> and [Tf<sub>2</sub>N]<sup>-</sup>, from AIMD and MD<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>; (e) conductivity of ionic liquid electrolyte containing Na<sup>+</sup>[Tf<sub>2</sub>N]<sup>-</sup> and Li<sup>+</sup>[Tf<sub>2</sub>N]<sup>-[26]</sup>; (f) snapshots of sodiated and lithiated NiO surfaces<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>; (g) schematic cartoons showing different reaction modes between sodiation and lithiation<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>Fig.4
... [27]Application of AIMD in electrolyte (a)-(c) Diffusion trajectories of Li atoms in AIMD<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>; (d) radial distribution function between Na<sup>+</sup>/Li<sup>+</sup> and [Tf<sub>2</sub>N]<sup>-</sup>, from AIMD and MD<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>; (e) conductivity of ionic liquid electrolyte containing Na<sup>+</sup>[Tf<sub>2</sub>N]<sup>-</sup> and Li<sup>+</sup>[Tf<sub>2</sub>N]<sup>-[26]</sup>; (f) snapshots of sodiated and lithiated NiO surfaces<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>; (g) schematic cartoons showing different reaction modes between sodiation and lithiation<sup>[<xref ref-type="bibr" rid="R27">27</xref>]</sup>Fig.4
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
... [31];(b) 离子液体电解液([C n mim][TFSI]和[C n mim][FSI], n = 2, 4)的密度和黏度与LiTFSI浓度的关系[31];(c) 有机溶剂电解液和离子液体电解液的电导率曲线[32];(d) [P13][OTf]-Li2S8 和[P13][TFSI]-Li2S8 中Li--S82- 和Li+-anions的质心RDF及配位数曲线[33]Application of MD in electrolyte (a) Snapshots of different lithium concentrations from MD<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (b) Density and viscosity <i>vs.</i> concentration of LiTFSI for all ionic liquid electrolytes ([C <i><sub>n</sub></i> mim][TFSI] and [C <i><sub>n</sub></i> mim][FSI], <i>n</i> = 2, 4)<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte<sup>[<xref ref-type="bibr" rid="R32">32</xref>]</sup>; (d) Center-of-mass RDFs and coordination number curves of Li<sup>+</sup>-S<sub>8</sub><sup>2-</sup> and Li<sup>+</sup>-anions for [P<sub>13</sub>][OTf]-Li<sub>2</sub>S<sub>8</sub>and [P<sub>13</sub>][TFSI]-Li<sub>2</sub>S<sub>8</sub> systems<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>Fig. 5
... [31];(c) 有机溶剂电解液和离子液体电解液的电导率曲线[32];(d) [P13][OTf]-Li2S8 和[P13][TFSI]-Li2S8 中Li--S82- 和Li+-anions的质心RDF及配位数曲线[33]Application of MD in electrolyte (a) Snapshots of different lithium concentrations from MD<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (b) Density and viscosity <i>vs.</i> concentration of LiTFSI for all ionic liquid electrolytes ([C <i><sub>n</sub></i> mim][TFSI] and [C <i><sub>n</sub></i> mim][FSI], <i>n</i> = 2, 4)<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte<sup>[<xref ref-type="bibr" rid="R32">32</xref>]</sup>; (d) Center-of-mass RDFs and coordination number curves of Li<sup>+</sup>-S<sub>8</sub><sup>2-</sup> and Li<sup>+</sup>-anions for [P<sub>13</sub>][OTf]-Li<sub>2</sub>S<sub>8</sub>and [P<sub>13</sub>][TFSI]-Li<sub>2</sub>S<sub>8</sub> systems<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>Fig. 5
... [31]; (b) Density and viscosity vs. concentration of LiTFSI for all ionic liquid electrolytes ([C n mim][TFSI] and [C n mim][FSI], n = 2, 4)[31]; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte[32]; (d) Center-of-mass RDFs and coordination number curves of Li+-S82- and Li+-anions for [P13][OTf]-Li2S8and [P13][TFSI]-Li2S8 systems[33]Fig. 5
... [31]; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte[32]; (d) Center-of-mass RDFs and coordination number curves of Li+-S82- and Li+-anions for [P13][OTf]-Li2S8and [P13][TFSI]-Li2S8 systems[33]Fig. 5
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
... [32];(d) [P13][OTf]-Li2S8 和[P13][TFSI]-Li2S8 中Li--S82- 和Li+-anions的质心RDF及配位数曲线[33]Application of MD in electrolyte (a) Snapshots of different lithium concentrations from MD<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (b) Density and viscosity <i>vs.</i> concentration of LiTFSI for all ionic liquid electrolytes ([C <i><sub>n</sub></i> mim][TFSI] and [C <i><sub>n</sub></i> mim][FSI], <i>n</i> = 2, 4)<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte<sup>[<xref ref-type="bibr" rid="R32">32</xref>]</sup>; (d) Center-of-mass RDFs and coordination number curves of Li<sup>+</sup>-S<sub>8</sub><sup>2-</sup> and Li<sup>+</sup>-anions for [P<sub>13</sub>][OTf]-Li<sub>2</sub>S<sub>8</sub>and [P<sub>13</sub>][TFSI]-Li<sub>2</sub>S<sub>8</sub> systems<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>Fig. 5
... [32]; (d) Center-of-mass RDFs and coordination number curves of Li+-S82- and Li+-anions for [P13][OTf]-Li2S8and [P13][TFSI]-Li2S8 systems[33]Fig. 5
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
... [33]Application of MD in electrolyte (a) Snapshots of different lithium concentrations from MD<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (b) Density and viscosity <i>vs.</i> concentration of LiTFSI for all ionic liquid electrolytes ([C <i><sub>n</sub></i> mim][TFSI] and [C <i><sub>n</sub></i> mim][FSI], <i>n</i> = 2, 4)<sup>[<xref ref-type="bibr" rid="R31">31</xref>]</sup>; (c) Conductivity of organic solvent electrolyte and ionic liquid electrolyte<sup>[<xref ref-type="bibr" rid="R32">32</xref>]</sup>; (d) Center-of-mass RDFs and coordination number curves of Li<sup>+</sup>-S<sub>8</sub><sup>2-</sup> and Li<sup>+</sup>-anions for [P<sub>13</sub>][OTf]-Li<sub>2</sub>S<sub>8</sub>and [P<sub>13</sub>][TFSI]-Li<sub>2</sub>S<sub>8</sub> systems<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>Fig. 5
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
1
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
1
... MD模拟是在足够长的时间内系统随时间的演化过程,产生足够多的满足需要的相空间的构型,然后对这些构型进行系综平均,可以有效得到体系的微观结构、动力学、热力学等宏观性质.例如本团队[31]采用MD模拟方法对4种离子液体电解液[LiTFSI-[C n mim][TFSI]和[C n mim][FSI],(n=2、4)]在不同锂盐浓度下的黏度、密度、锂离子迁移数、自扩散系数等性质进行了探究[图5(a)、(b)].研究表明,所有离子液体电解液的密度和黏度都随着锂盐浓度的增加而增加,且模拟结果与实验数值高度吻合.同时,发现锂盐浓度的增加可使锂离子与离子液体阴离子的配位结构更加紧密,从而形成团簇结构促进锂离子迁移.随后,本团队[32]又通过MD计算对比了传统有机溶剂电解液(LiTFSI-DMC、DEC)和离子液体电解液[(LiTFSI-[C n mim][TFSI]、[C n mim][BF4](n=2,4)]在高浓度下的溶剂化结构.如图5(c)所示,有机溶剂在高锂盐浓度下极大限制了离子自由运动,导致体系的电导率降低.此外,由于MD计算可以获得所有原子实时的运动轨迹,通过分析可以直接获得离子的扩散系数,常用于研究电解液的离子扩散行为及扩散机制.因此,本团队的Hu等[33]通过MD模拟探究了Li2S8在离子液体和离子液体基电解液中的微观机制和传输行为.图5(d)显示,相比于[TFSI]-,[OTF] -与Li+具有更高的配位强度.并且[OTF]-可以加速电解液中的Li+交换速率,使Li+溶剂化层更容易分解.同年,Hu等[34]更详细地研究了不同[PP13]基离子液体与短链多硫化物的聚集行为,离子液体与短链聚合物容易形成一种“三明治”结构.相似地,Kirchner等[35]通过对比4种离子液体电解液,进一步得出Li+迁移率和输运与离子液体相互作用正相关并且与Li+溶解率成反比. ...
... [37];[Pyr13][TFSI]/MeTFSI (Me= Li+ 、Ni2+ 等)中的(c)离子团簇和(d)团簇内的结合能[38](a) two Li<sup>+</sup> cation solvation structures diagrammatic drawing and (b) Li<sup>+</sup> contribution to ionic conductivity of ionic liquid electrolytes ([Pyr<sub>13</sub>][TFSI]/LiTFSI)<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>; (c) representative ion clusters from small MD simulations and (d) binding energies of ion clusters<sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig. 6
(a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
(a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
... [38](a) two Li<sup>+</sup> cation solvation structures diagrammatic drawing and (b) Li<sup>+</sup> contribution to ionic conductivity of ionic liquid electrolytes ([Pyr<sub>13</sub>][TFSI]/LiTFSI)<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>; (c) representative ion clusters from small MD simulations and (d) binding energies of ion clusters<sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig. 6
(a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
(a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
... [40];[Pyr14][TFSI]/MeTFSI(Me=Li+ 、Ni2+ 等)中[pyr14]的(c)均方位移对数表示曲线和(d)自扩散系数[41](a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
... [40]; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr14] of [Pyr14][TFSI] / MeTFSI (Me= Li+, Ni2+)[41]Fig. 7
... [41](a) metal cation residence time and (b) displacement of metal cations during residence time of [Pyr<sub>14</sub>][TFSI]/MeTFSI (Me= Li<sup>+</sup>, Na<sup>+</sup>, Mg<sup>+</sup>, Zn<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) logarithmic representation of mean squared and (d) self-diffusion coefficients for [pyr<sub>14</sub>] of [Pyr<sub>14</sub>][TFSI] / MeTFSI (Me= Li<sup>+</sup>, Ni<sup>2+</sup>)<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>Fig. 7
... [64];(b) 离子液体和有机电解液的积分电容与孔隙大小的关系[65];(c) 由中孔和微孔组成的结构模型,孔内模拟快照及[Bmim]和[TFSI]的离子密度分布[66];(d) 孔内阳离子和阴离子在电极上的分布[67]Simulation of ionic liquid electrolytes in porous electrode capacitor: (a) influence of electrode curvature on differential capacitance<sup>[<xref ref-type="bibr" rid="R64">64</xref>]</sup>; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte<sup>[<xref ref-type="bibr" rid="R65">65</xref>]</sup>; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI]<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (d) in-pore cation and anion population at electrodes<sup>[<xref ref-type="bibr" rid="R67">67</xref>]</sup>Fig. 9
... [64]; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte[65]; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI][66]; (d) in-pore cation and anion population at electrodes[67]Fig. 9
... [65];(c) 由中孔和微孔组成的结构模型,孔内模拟快照及[Bmim]和[TFSI]的离子密度分布[66];(d) 孔内阳离子和阴离子在电极上的分布[67]Simulation of ionic liquid electrolytes in porous electrode capacitor: (a) influence of electrode curvature on differential capacitance<sup>[<xref ref-type="bibr" rid="R64">64</xref>]</sup>; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte<sup>[<xref ref-type="bibr" rid="R65">65</xref>]</sup>; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI]<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (d) in-pore cation and anion population at electrodes<sup>[<xref ref-type="bibr" rid="R67">67</xref>]</sup>Fig. 9
... [65]; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI][66]; (d) in-pore cation and anion population at electrodes[67]Fig. 9
... [66];(d) 孔内阳离子和阴离子在电极上的分布[67]Simulation of ionic liquid electrolytes in porous electrode capacitor: (a) influence of electrode curvature on differential capacitance<sup>[<xref ref-type="bibr" rid="R64">64</xref>]</sup>; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte<sup>[<xref ref-type="bibr" rid="R65">65</xref>]</sup>; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI]<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (d) in-pore cation and anion population at electrodes<sup>[<xref ref-type="bibr" rid="R67">67</xref>]</sup>Fig. 9
Simulation of ionic liquid electrolytes in porous electrode capacitor: (a) influence of electrode curvature on differential capacitance<sup>[<xref ref-type="bibr" rid="R64">64</xref>]</sup>; (b) dependence of integral capacitance of an EDLC on pore size for ionic liquid and organic electrolyte<sup>[<xref ref-type="bibr" rid="R65">65</xref>]</sup>; (c) structural model showing mesopore and micropore components, simulation snapshots and ion density distributions of [Bmim] and [TFSI]<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (d) in-pore cation and anion population at electrodes<sup>[<xref ref-type="bibr" rid="R67">67</xref>]</sup>Fig. 9




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}