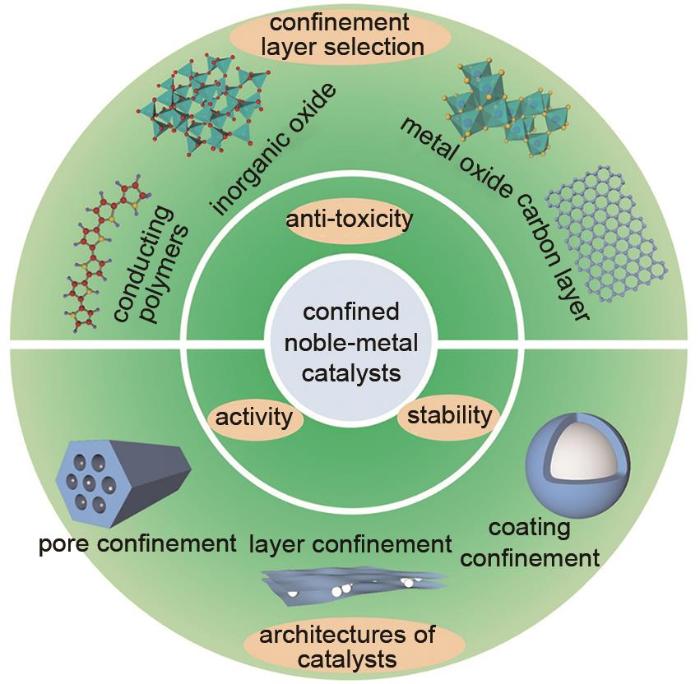
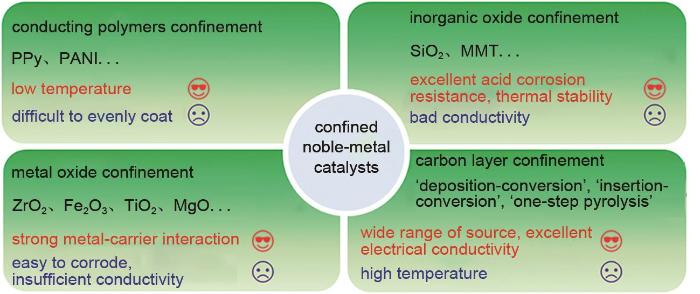
Developing highly efficient oxygen reduction reaction (ORR) electrocatalysts is one of the main techniques to achieve the large-scale application of proton exchange membrane fuel cells (PEMFCs). Noble-metal based catalysts, as robust ORR catalysts, are the most likely candidates for practical application. The large-scale use of noble metal may lead to a high cost of the catalysts. On the other hand, the stabilities of noble-metal catalysts still need further improvements. Confining noble-metal catalysts in physical confinement layer can greatly improve catalytic stability without compromising initial activity, thus prolonging the service life of catalysts. The physical confinement layers can not only inhibit the coalescence of catalysts during high-temperature preparation, but also mitigate the aggregation, detachment and dissolution during electrochemical process. In this review, confined noble-metal ORR catalysts are reviewed, including conducting polymer confined noble-metal catalysts, non-metal-oxide confined noble-metal catalysts, metal-oxides confined catalysts and carbon confined catalysts. Besides, the structure-performance relationship between confinement layers' physical properties and electrocatalytic performance is analyzed. Three main strategies to achieve carbon confinement are emphasized, including 'deposition-conversion' strategy, 'insertion-conversion' strategy and 'one-step pyrolysis' strategy. At last, summary and prospect are given, also some existing challenges are stated.
HU Yezhou. Recent progress in confined noble-metal electrocatalysts for oxygen reduction reaction[J]. Energy Storage Science and Technology, 2022, 11(4): 1264-1277
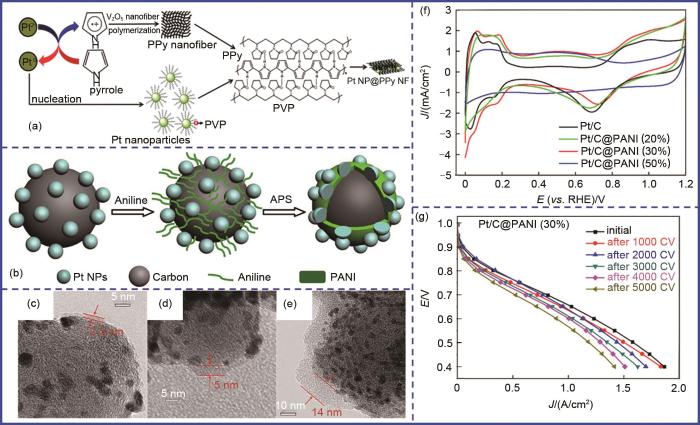
Fig. 2
Conducting polymers confinement. (a) Schematic diagram of Pt NP@PPy NF; (b) Schematic diagram of Pt/C@PANI; (c)—(e) TEM images and (f) CV curves of Pt/C@PANI with different thickness of coating layer; (g) MEA stability test of Pt/C@PANI (30%)
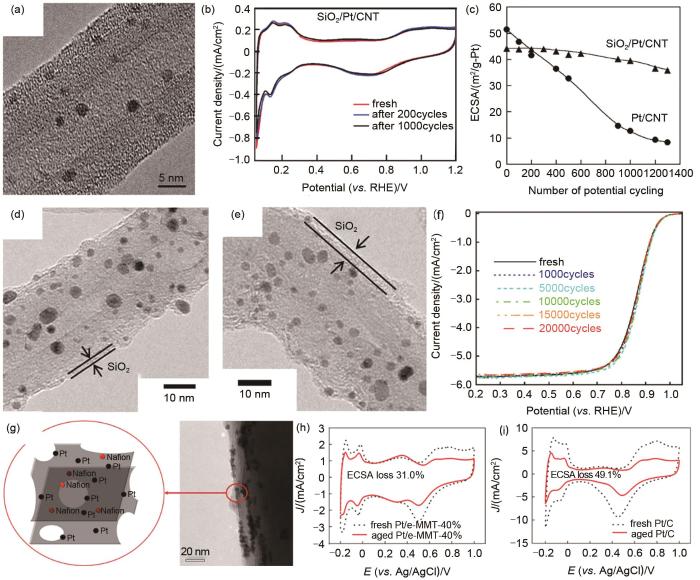
Fig. 3
Inorganic oxide confinement. (a) TEM image of SiO2/Pt/CNT using TEOS as precursor; (b) CV curves and (c)ECSA change of SiO2/Pt/CNT at different potential cycles, (d)—(e) TEM images of SiO2/Pt/CNT(MTEOS) using MTEOS as precursor; (f) ORR stability test of SiO2/Pt/CNT(MTEOS); (g) TEM image of Pt/e-MMT; The CV curves of (h) Pt/e-MMT and (i) Pt/C before and after stability tests
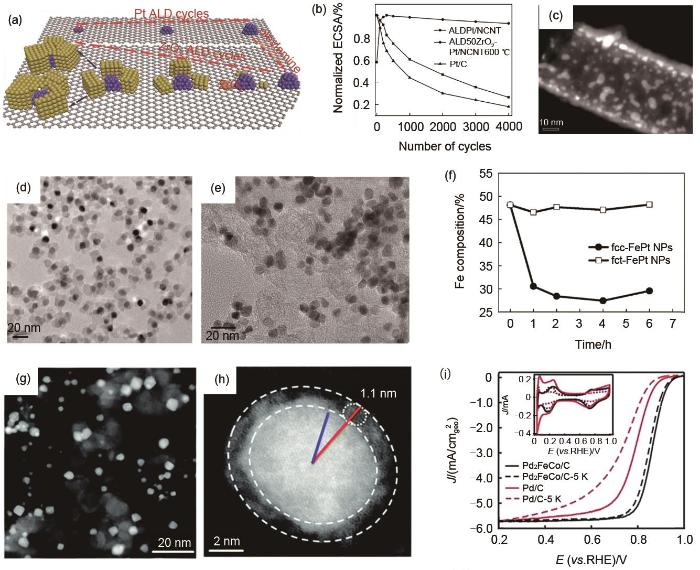
Fig. 4
Metal oxides confinement. (a) Schematic diagram of ZrO2 confined Pt nanoparticles; (b) The normalized ECSA of ALDPt/NCNT, ALD50ZrO2-Pt/NCNT600 ℃ and E-TEK Pt/CPMTT at different cycling stages; (c) The STEM image of ALD50ZrO2-Pt/NCNT after stability test; (d) MgO confined fcc-FePt nanoparticles; (e) fct-FePt nanoparticles after removal of MgO; (f) The Fe composition in fcc-FePt/C and fct-FePt/C at different times; (g)—(h) STEM images of Fe2O3 confined Pd2FeCo nanoparticles; (i) The stability test of Pd2FeCo/C and Pd/C
Fig. 5
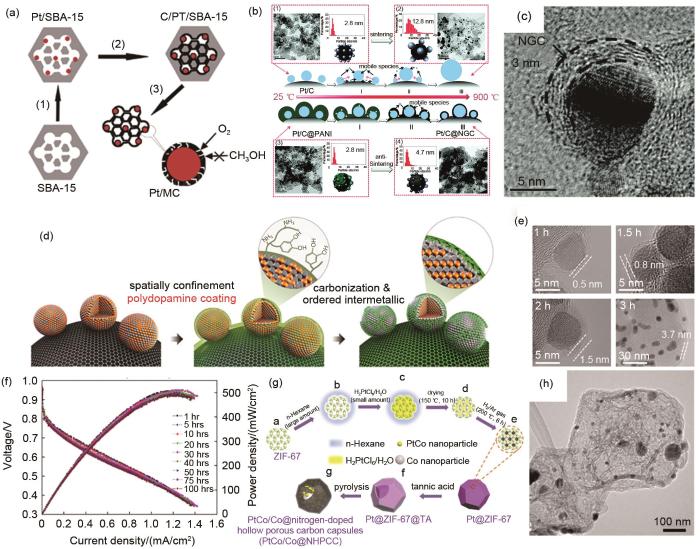
Carbon confinement base on ‘deposition-conversion’ strategy. (a) Schematic diagram of Pt@C/MC; (b) Schematic diagram of Pt/C@NGC; (c) The TEM image of Pt/C@NGC; (d) Schematic diagram of L10-PtFe nanoparticles; (e) The TEM images of L10-PtFe nanoparticles with different thickness of carbon layers; (f) The stability test of carbon confined L10-PtFe nanoparticles in MEA; (g) Schematic diagram of PtCo/Co@NHPCC; (h) The TEM image of PtCo/Co@NHPCC
Fig. 6
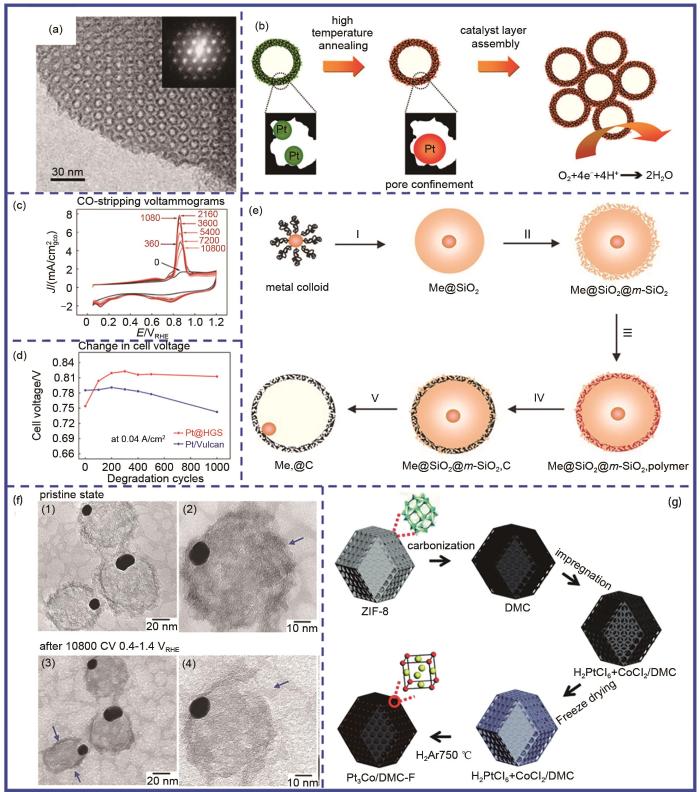
Carbon confinement base on ‘insertion-conversion’ strategy. (a) The TEM image of ordered nanoporous arrays of carbon; (b) The schematic diagram of Pt@HGS; (c) The CO stripping curves of Pt@HSG at different potential cycling; (d) Comparison of cell voltage change of Pt@HGS and Pt/Vulcan during stability test; (e) Schematic diagram of encapsulated metal nanoparticles in porous carbon shells; (f) IL-TEM images of AuPt@C before and after stability test; (g) Schematic diagram of Pt3Co/DMF-F
Fig. 7
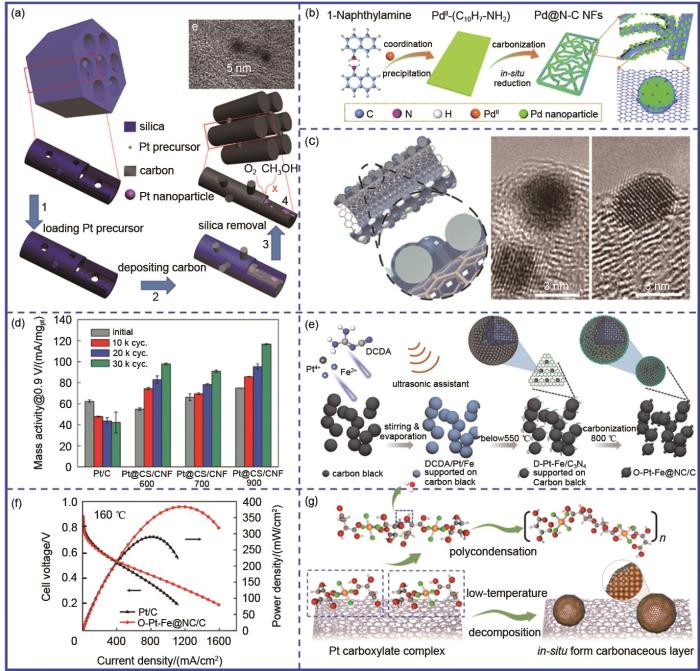
Carbon confinement base on ‘one-step pyrolysis’ strategy. (a) The schematic diagram of Pt@GC; (b) The schematic diagram of Pd@N-C NFs; (c) The schematic diagram of Pt@CN x /CNT; (d) The stability tests of Pt@CS/CNF; (e) The schematic diagram of O-Pt-Fe@NC/C; (f) The high-temperature fuel cell polarization curves of O-Pt-Fe@NC/C and Pt/C; (g) The synthesis of Pt@C/C trough one-step low temperature pyrolysis
Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)]。热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆。其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触。其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题。萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能。Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)]。Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减。这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等。类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF)。通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构。Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)]。一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金。结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)]。利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C)。考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性。得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善。
HAN A L, ZHANG Z D, YANG J R, et al. Carbon-supported single-atom catalysts for formic acid oxidation and oxygen reduction reactions[J]. Small, 2021, 17(16): doi: 10.1002/smll.202004500.
QIAO Z, HWANG S, LI X, et al. 3D porous graphitic nanocarbon for enhancing the performance and durability of Pt catalysts: A balance between graphitization and hierarchical porosity[J]. Energy & Environmental Science, 2019, 12(9): 2830-2841.
HAIDER R, WEN Y C, MA Z F, et al. High temperature proton exchange membrane fuel cells: Progress in advanced materials and key technologies[J]. Chemical Society Reviews, 2021, 50(2): 1138-1187.
GAO W B, LIU T T, ZHANG Z P, et al. Stabilization of Pt nanoparticles at the Ta2O5-TaC binary junction: An effective strategy to achieve high durability for oxygen reduction[J]. Journal of Materials Chemistry A, 2020, 8(11): 5525-5534.
SHI Q R, ZHU C Z, DU D, et al. Robust noble metal-based electrocatalysts for oxygen evolution reaction[J]. Chemical Society Reviews, 2019, 48(12): 3181-3192.
HU Y Z, LU Y, ZHAO X R, et al. Highly active N-doped carbon encapsulated Pd-Fe intermetallic nanoparticles for the oxygen reduction reaction[J]. Nano Research, 2020, 13(9): 2365-2370.
TIAN N, ZHOU Z Y, SUN S G, et al. Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity[J]. Science, 2007, 316(5825): 732-735.
WANG G Z, YANG Z Z, DU Y G, et al. Programmable exposure of Pt active facets for efficient oxygen reduction[J]. Angewandte Chemie International Edition, 2019, 58(44): 15848-15854.
YANG Y, XIAO W P, FENG X R, et al. Golden palladium zinc ordered intermetallics as oxygen reduction electrocatalysts[J]. ACS Nano, 2019, 13(5): 5968-5974.
KONG Z J, MASWADEH Y, VARGAS J A, et al. Origin of high activity and durability of twisty nanowire alloy catalysts under oxygen reduction and fuel cell operating conditions[J]. Journal of the American Chemical Society, 2020, 142(3): 1287-1299.
GONG M X, DENG Z P, XIAO D D, et al. One-nanometer-thick Pt3Ni bimetallic alloy nanowires advanced oxygen reduction reaction: Integrating multiple advantages into one catalyst[J]. ACS Catalysis, 2019, 9(5): 4488-4494.
BU L Z, ZHANG N, GUO S J, et al. Biaxially strained PtPb/Pt core/shell nanoplate boosts oxygen reduction catalysis[J]. Science, 2016, 354(6318): 1410-1414.
GONG M X, XIAO D D, DENG Z P, et al. Structure evolution of PtCu nanoframes from disordered to ordered for the oxygen reduction reaction[J]. Applied Catalysis B: Environmental, 2021, 282: doi: 10.1016/j.apcatb.2020.119617.
ZANA A, SPEDER J, REELER N E A, et al. Investigating the corrosion of high surface area carbons during start/stop fuel cell conditions: A Raman study[J]. Electrochimica Acta, 2013, 114: 455-461.
YANG Z, BALL S, CONDIT D, et al. Systematic study on the impact of Pt particle size and operating conditions on PEMFC cathode catalyst durability[J]. Journal of the Electrochemical Society, 2011, 158(11): B1439.
LIU Y, LU N, POYRAZ S, et al. One-pot formation of multifunctional Pt-conducting polymer intercalated nanostructures[J]. Nanoscale, 2013, 5(9): 3872-3879.
MIZUHATA M, OGA M, DEKI S. Preparation of Pt/polypyrrole loaded carbon composite in order to improve electrode durability for fuel cells[J]. ECS Transactions, 2007, 2(8): 63-72.
CHEN S G, WEI Z D, QI X Q, et al. Nanostructured polyaniline-decorated Pt/C@PANI core-shell catalyst with enhanced durability and activity[J]. Journal of the American Chemical Society, 2012, 134(32): 13252-13255.
TAKENAKA S, MATSUMORI H, NAKAGAWA K, et al. Improvement in the durability of Pt electrocatalysts by coverage with silica layers[J]. The Journal of Physical Chemistry C, 2007, 111(42): 15133-15136.
TAKENAKA S, MIYAMOTO H, UTSUNOMIYA Y, et al. Catalytic activity of highly durable Pt/CNT catalysts covered with hydrophobic silica layers for the oxygen reduction reaction in PEFCs[J]. The Journal of Physical Chemistry C, 2014, 118(2): 774-783.
AOKI N, INOUE H, KAWASAKI H, et al. Durability improvement of Pd core-Pt shell structured catalyst by porous SiO2 coating[J]. Journal of the Electrochemical Society, 2018, 165(10): F737-F747.
LI W, DING W, NIE Y, et al. Enhancing the stability and activity by anchoring Pt nanoparticles between the layers of etched montmorillonite for oxygen reduction reaction[J]. Science Bulletin, 2016, 61(18): 1435-1439.
ANDO F, GUNJI T K, TANABE T, et al. Enhancement of the oxygen reduction reaction activity of Pt by tuning its d-band center via transition metal oxide support interactions[J]. ACS Catalysis, 2021, 11(15): 9317-9332.
CHENG N C, BANIS M N, LIU J, et al. Extremely stable platinum nanoparticles encapsulated in a zirconia nanocage by area-selective atomic layer deposition for the oxygen reduction reaction[J]. Advanced Materials, 2015, 27(2): 277-281.
KANG S, XIA F, HU Z F, et al. Platinum nanoparticles with TiO2-skin as a durable catalyst for photoelectrochemical methanol oxidation and electrochemical oxygen reduction reactions[J]. Electrochimica Acta, 2020, 343: doi: 10.1016/j.electacta.2020.136119.
KIM J, LEE Y, SUN S H. Structurally ordered FePt nanoparticles and their enhanced catalysis for oxygen reduction reaction[J]. Journal of the American Chemical Society, 2010, 132(14): 4996-4997.
XIAO W P, LIUTHEVICIENE CORDEIRO M A, GONG M X, et al. Optimizing the ORR activity of Pd based nanocatalysts by tuning their strain and particle size[J]. Journal of Materials Chemistry A, 2017, 5(20): 9867-9872.
WEN Z, LIU J, LI J. Core/shell Pt/C nanoparticles embedded in mesoporous carbon as a methanol-tolerant cathode catalyst in direct methanol fuel cells[J]. Advanced Materials, 2008, 20(4): 743-747.
NIE Y, CHEN S G, DING W, et al. Pt/C trapped in activated graphitic carbon layers as a highly durable electrocatalyst for the oxygen reduction reaction[J]. Chemical Communications (Cambridge, England), 2014, 50(97): 15431-15434.
TAN J L, XIE Z, ZHANG Z, et al. Dopamine modified polyaniline with improved adhesion, dispersibility, and biocompatibility[J]. Journal of Materials Science, 2018, 53(1): 447-455.
CHUNG D Y, JUN S W, YOON G, et al. Highly durable and active PtFe nanocatalyst for electrochemical oxygen reduction reaction[J]. Journal of the American Chemical Society, 2015, 137(49): 15478-15485.
SUN K, LI J, WANG F, et al. Highly enhanced durability of a graphitic carbon layer decorated PtNi3 alloy electrocatalyst toward the oxygen reduction reaction[J]. Chemical Communications (Cambridge, England), 2019, 55(40): 5693-5696.
YING J, LI J, JIANG G P, et al. Metal-organic frameworks derived platinum-cobalt bimetallic nanoparticles in nitrogen-doped hollow porous carbon capsules as a highly active and durable catalyst for oxygen reduction reaction[J]. Applied Catalysis B: Environmental, 2018, 225: 496-503.
JOO S H, CHOI S J, OH I, et al. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles[J]. Nature, 2001, 412 (6843): 169-172.
GALEANO C, MEIER J C, PEINECKE V, et al. Toward highly stable electrocatalysts via nanoparticle pore confinement[J]. Journal of the American Chemical Society, 2012, 134(50): 20457-20465.
ZHAO W Y, YE Y K, JIANG W J, et al. Mesoporous carbon confined intermetallic nanoparticles as highly durable electrocatalysts for the oxygen reduction reaction[J]. Journal of Materials Chemistry A, 2020, 8(31): 15822-15828.
WU Z X, LV Y Y, XIA Y Y, et al. Ordered mesoporous platinum@graphitic carbon embedded nanophase as a highly active, stable, and methanol-tolerant oxygen reduction electrocatalyst[J]. Journal of the American Chemical Society, 2012, 134(4): 2236-2245.
JIANG X, WANG J X, HUANG T, et al. Sub-5 nm palladium nanoparticles in situ embedded in N-doped carbon nanoframes: Facile synthesis, excellent sinter resistance and electrocatalytic properties[J]. Journal of Materials Chemistry A, 2019, 7(46): 26243-26249.
KARUPPANNAN M, KIM Y, GOK S, et al. A highly durable carbon-nanofiber-supported Pt-C core-shell cathode catalyst for ultra-low Pt loading proton exchange membrane fuel cells: Facile carbon encapsulation[J]. Energy & Environmental Science, 2019, 12(9): 2820-2829.
HU Y Z, SHEN T, ZHAO X R, et al. Combining structurally ordered intermetallics with N-doped carbon confinement for efficient and anti-poisoning electrocatalysis[J]. Applied Catalysis B: Environmental, 2020, 279: doi: 10.1016/j.apcatb.2020.119370.
HU Y Z, ZHANG J J, SHEN T, et al. A low-temperature carbon encapsulation strategy for stable and poisoning-tolerant electrocatalysts[J]. Small Methods, 2021, 5(11): doi: 10.1002/smtd.202100937.
... Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)].热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆.其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触.其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题.萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能.Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)].Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减.这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等.类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF).通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构.Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)].一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金.结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)].利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C).考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性.得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善. ...
... Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)].热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆.其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触.其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题.萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能.Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)].Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减.这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等.类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF).通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构.Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)].一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金.结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)].利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C).考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性.得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善. ...
1
... Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)].热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆.其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触.其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题.萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能.Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)].Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减.这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等.类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF).通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构.Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)].一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金.结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)].利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C).考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性.得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善. ...
1
... Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)].热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆.其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触.其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题.萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能.Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)].Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减.这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等.类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF).通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构.Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)].一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金.结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)].利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C).考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性.得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善. ...
1
... Lee等[40]利用有机配体与Pd离子之间的配位作用,先通过1-萘胺与Pd配位形成层状配位聚合物,然后将得到的配位聚合物直接碳化得到具有框架结构的碳包覆Pd催化剂(Pd@N-C NFs)[图7(b)].热解过程中,层状Pd-NA前驱体转变为多孔框架结构,Pd以约5.5 nm粒径均匀分布在碳骨架上且被较薄的碳层所包覆.其中1-萘胺作为氮源和碳源,不仅有利于氮掺杂碳的生成,更有利于金属催化剂与氮掺杂碳基底之间实现更紧密的接触.其次金属离子与含氮配体之间的相互作用也有利于防止热处理过程中颗粒的团聚、长大等问题.萘胺中杂原子掺杂有利于产生多功能杂原子掺杂碳,进一步提升电催化性能.Wan等[41]以(NH4)2S2O8为引发剂,PANI为碳源与H2PtCl6和CNT在低温条件下进行聚合,并通过后续高温碳化、酸洗得到氮掺杂多孔碳限域的Pt纳米颗粒(Pt@CN x /CNT)[图7(c)].Pt@CN x /CNT在酸性条件下循环1500圈以后,半波电位几乎没有发生明显衰减.这主要是由于氮掺杂碳层对Pt纳米颗粒的限域作用避免了其在载体上的脱落、团聚等.类似地,Kwon等[42]将Pt-苯胺配位化合物与CNF进行均匀混合,通过后续一步高温热处理得到CNF负载的碳层限域Pt纳米颗粒(Pt@CS/CNF).通过改变碳化温度对碳层石墨化程度进行调控发现,900 ℃处理后的Pt@CS/CNF展现出最优的ORR稳定性[图7(d)],意味着除了对碳层厚度进行合理调控外,还应关注其石墨化结构.Wang等[43]以双氰胺作为碳层前驱体,与H2PtCl6、FeCl2及Vulcan碳黑均匀混合后,通过一步高温热处理得到了超薄氮掺杂碳限域的PtFe有序合金(O-PtFe@NC/C)[图7(e)].一步高温热处理中双氰胺先在较低温下转变为石墨化C3N4,随着温度的进一步升高,C3N4进行分解,并完成在PtFe合金表面的原位碳层包覆,同时无序PtFe进一步转变为有序PtFe合金.结果表明,碳层限域一方面抑制了合金高温有序化过程中的颗粒团聚问题,还能够有效抑制毒化物种如SO x 、PO x 等在催化剂表面的吸附,从而提高催化剂的抗毒化性能,在160 ℃下,O-Pt-Fe@NC/C组装的高温质子交换膜燃料电池的最大功率密度高达384 mW/cm2 [图7(f)].利用类似的方法,Wang等[6]实现了多孔氮掺杂碳限域的有序Pd-Fe合金催化剂(O-Pd-Fe@NC/C).考虑到Pd相较于Pt具有相对较低的氧化还原电位(Pd: 0.951 V,Pt: 1.18 V),我们发现碳限域层需要达到相对较厚的状态才能够一定程度上提高Pd基催化剂的ORR稳定性.得益于碳层限域作用,所得O-Pd-Fe@NC/C催化剂的MA衰减程度明显降低,且抗CO及SO x 性能也得到明显改善. ...




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}