[1]
CHU S, MAJUMDAR A. Opportunities and challenges for a sustainable energy future[J]. Nature, 2012, 488(7411): 294-303.
[本文引用: 1]
[2]
SEH Z W, KIBSGAARD J, DICKENS C F, et al. Combining theory and experiment in electrocatalysis: Insights into materials design[J]. Science, 2017, 355(6321): eaad4998.
[本文引用: 4]
[3]
LARCHER D, TARASCON J M. Towards greener and more sustainable batteries for electrical energy storage[J]. Nature Chemistry, 2015, 7(1): 19-29.
[本文引用: 1]
[4]
STAMENKOVIC V R, STRMCNIK D, LOPES P P, et al. Energy and fuels from electrochemical interfaces[J]. Nature Materials, 2017, 16(1): 57-69.
[本文引用: 1]
[5]
国家能源局. 国家能源局举行新闻发布会介绍2021年上半年能源经济形势等情况 [R/OL]. [2021-07-29]. http://wwwneagovcn/2021-07/29/c_1310093667htm.
[本文引用: 1]
[6]
TANG C, WANG H F, ZHANG Q. Multiscale principles to boost reactivity in gas-involving energy electrocatalysis[J]. Accounts of Chemical Research, 2018, 51(4): 881-889.
[本文引用: 1]
[7]
JAKA Tušek, KURT Engelbrecht, Dan ERIKSEN, et al. A regenerative elastocaloric heat pump[J]. Nature Energy, 2016, 74(1): 16134.
[8]
GLENK G, REICHELSTEIN S. Economics of converting renewable power to hydrogen[J]. Nature Energy, 2019, 4(3): 216-222.
[本文引用: 1]
[9]
DOTAN H, LANDMAN A, SHEEHAN S W, et al. Decoupled hydrogen and oxygen evolution by a two-step electrochemical-chemical cycle for efficient overall water splitting[J]. Nature Energy, 2019, 4(9): 786-795.
[本文引用: 1]
[10]
SYMES M D, CRONIN L. Decoupling hydrogen and oxygen evolution during electrolytic water splitting using an electron-coupled-proton buffer[J]. Nature Chemistry, 2013, 5(5): 403-409.
[本文引用: 1]
[11]
VAN H N. The clean hydrogen future has already begun [EB/OL] [2019-04-23]. https://wwwieaorg/newsroom/news/2019/april/the-clean-hydrogen-future-has-already-begunhtml.
[本文引用: 1]
[12]
LAGADEC M F, GRIMAUD A. Water electrolysers with closed and open electrochemical systems[J]. Nature Materials, 2020, 19(11): 1140-1150.
[本文引用: 1]
[13]
中国氢能联盟. 中国氢能源及燃料电池产业白皮书 [R/OL]. [2020-02-20]. http://wwwtanjiaoyicom/article-27580-1html.
[本文引用: 1]
[14]
GROCHALA W. First there was hydrogen[J]. Nature Chemistry, 2015, 7(3): 264.
[本文引用: 1]
[15]
ZOU X X, ZHANG Y. Noble metal-free hydrogen evolution catalysts for water splitting[J]. Chemical Society Reviews, 2015, 44(15): 5148-5180.
[本文引用: 3]
[16]
CONWAY B E, TILAK B V. Interfacial processes involving electrocatalytic evolution and oxidation of H2 , and the role of chemisorbed H[J]. Electrochimica Acta, 2002, 47(22/23): 3571-3594.
[本文引用: 2]
[17]
DUBOUIS N, GRIMAUD A. The hydrogen evolution reaction: From material to interfacial descriptors[J]. Chemical Science, 2019, 10(40): 9165-9181.
[本文引用: 1]
[18]
YU X W, ZHAO J, ZHENG L R, et al. Hydrogen evolution reaction in alkaline media: Alpha- or beta-nickel hydroxide on the surface of platinum? [J]. ACS Energy Letters, 2018, 3(1): 237-244.
[本文引用: 1]
[19]
WANG J, XU F, JIN H Y, et al. Non-noble metal-based carbon composites in hydrogen evolution reaction: Fundamentals to applications[J]. Advanced Materials, 2017, 29(14): doi:10.1002/adma.201605838.
[本文引用: 1]
[20]
SULTAN S, TIWARI J N, SINGH A N, et al. Single atoms and clusters based nanomaterials for hydrogen evolution, oxygen evolution reactions, and full water splitting[J]. Advanced Energy Materials, 2019, 9(22): doi:10.1002/aenm.201900624.
[本文引用: 1]
[21]
SUN H M, YAN Z H, LIU F M, et al. Self-supported transition-metal-based electrocatalysts for hydrogen and oxygen evolution[J]. Advanced Materials, 2020, 32(3): doi:10.1002/adma.201806326.
[本文引用: 7]
[22]
MAHMOOD N, YAO Y D, ZHANG J W, et al. Electrocatalysts for hydrogen evolution in alkaline electrolytes: Mechanisms, challenges, and prospective solutions[J]. Advanced Science, 2018, 5(2): doi:10.1002/advs.201700464.
[本文引用: 1]
[23]
WANG M, ZHANG L, HE Y J, et al. Recent advances in transition-metal-sulfide-based bifunctional electrocatalysts for overall water splitting[J]. Journal of Materials Chemistry A, 2021, 9(9): 5320-5363.
[本文引用: 5]
[24]
MORALES-GUIO C G, STERN L A, HU X L. Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution[J]. Chemical Society Reviews, 2014, 43(18): 6555-6569.
[本文引用: 3]
[25]
SANTOS D M F, SEQUEIRA C A C, FIGUEIREDO J L. Hydrogen production by alkaline water electrolysis[J]. Química Nova, 2013, 36(8): 1176-1193.
[本文引用: 1]
[26]
OJHA K, SAHA S M, DAGAR P, et al. Nanocatalysts for hydrogen evolution reactions[J]. Physical Chemistry Chemical Physics: PCCP, 2018, 20(10): 6777-6799.
[本文引用: 2]
[27]
SUEN N T, HUNG S F, QUAN Q, et al. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives[J]. Chemical Society Reviews, 2017, 46(2): 337-365.
[本文引用: 1]
[28]
LI D G, PARK E J, ZHU W L, et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers[J]. Nature Energy, 2020, 5(5): 378-385.
[本文引用: 2]
[29]
WANG P C, JIA T, WANG B G. A critical review: 1D/2D nanostructured self-supported electrodes for electrochemical water splitting[J]. Journal of Power Sources, 2020, 474: doi:10.1016/j.jpowsour.2020.228621.
[本文引用: 2]
[30]
SUN H M, XU X B, YAN Z H, et al. Superhydrophilic amorphous Co–B–P nanosheet electrocatalysts with Pt-like activity and durability for the hydrogen evolution reaction[J]. Journal of Materials Chemistry A, 2018, 6(44): 22062-22069.
[本文引用: 1]
[31]
LI J, ZHENG G F. One-dimensional earth-abundant nanomaterials for water-splitting electrocatalysts[J]. Advanced Science, 2017, 4(3): doi:10.1002/advs.201600380.
[本文引用: 1]
[32]
YAN Z H, SUN H M, CHEN X, et al. Anion insertion enhanced electrodeposition of robust metal hydroxide/oxide electrodes for oxygen evolution[J]. Nature Communications, 2018, 9: 2373.
[本文引用: 2]
[33]
WANG P C, JIA T, WANG B G. Review—recent advance in self-supported electrocatalysts for rechargeable zinc-air batteries[J]. Journal of the Electrochemical Society, 2020, 167(11): doi:10.1149/1945-7111/aba96e.
[本文引用: 2]
[34]
WAN L, WANG P C. Recent progress on self-supported two-dimensional transition metal hydroxides nanosheets for electrochemical energy storage and conversion[J]. International Journal of Hydrogen Energy, 2021, 46(12): 8356-8376.
[本文引用: 1]
[35]
WANG P C, WANG B G. Interface engineering of binder-free earth-abundant electrocatalysts for efficient advanced energy conversion[J]. ChemSusChem, 2020, 13(18): 4795-4811.
[本文引用: 3]
[36]
YANG H Y, DRIESS M, MENEZES P W. Self-supported electrocatalysts for practical water electrolysis[J]. Advanced Energy Materials, 2021, 11(39): doi:10.1002/aenm.202170153.
[本文引用: 2]
[37]
WANG P C, WANG B G. C-O-Co bond-stabilized CoP on carbon cloth toward hydrogen evolution reaction[J]. International Journal of Hydrogen Energy, 2022, 47(15): 9209-9219.
[本文引用: 1]
[38]
WANG P C, WANG R Z, XU Q, et al. Role of the interfacial effect between the substrate and Co(OH)2 layer in electrochemical oxygen evolution[J]. ACS Applied Energy Materials, 2021, 4(9): 9487-9497.
[本文引用: 3]
[39]
CLIMENT M J, CORMA A, IBORRA S. Heterogeneous catalysts for the one-pot synthesis of chemicals and fine chemicals[J]. Chemical Reviews, 2011, 111(2): 1072-1133.
[本文引用: 1]
[40]
KIM J, SOMORJAI G A. Molecular packing of lysozyme, fibrinogen, and bovine serum albumin on hydrophilic and hydrophobic surfaces studied by infrared-visible sum frequency generation and fluorescence microscopy[J]. Journal of the American Chemical Society, 2003, 125(10): 3150-3158.
[41]
BASHYAM R, ZELENAY P. A class of non-precious metal composite catalysts for fuel cells[J]. Nature, 2006, 443(7107): 63-66.
[本文引用: 1]
[42]
SUBBARAMAN R, TRIPKOVIC D, CHANG K C, et al. Trends in activity for the water electrolyser reactions on 3d M(Ni, Co, Fe, Mn) hydr(oxy)oxide catalysts[J]. Nature Materials, 2012, 11(6): 550-557.
[本文引用: 1]
[43]
ZHANG B, LIU J, WANG J S, et al. Interface engineering: The Ni(OH)2 /MoS2 heterostructure for highly efficient alkaline hydrogen evolution[J]. Nano Energy, 2017, 37: 74-80.
[本文引用: 3]
[44]
NIU S Q, SUN Y C, SUN G J, et al. Stepwise electrochemical construction of FeOOH/Ni(OH)2 on Ni foam for enhanced electrocatalytic oxygen evolution[J]. ACS Applied Energy Materials, 2019, 2(5): 3927-3935.
[本文引用: 1]
[45]
HU J, ZHANG C X, JIANG L, et al. Nanohybridization of MoS2 with layered double hydroxides efficiently synergizes the hydrogen evolution in alkaline media[J]. Joule, 2017, 1(2): 383-393.
[本文引用: 3]
[46]
LYU Q L, YANG L, WANG W, et al. One-step construction of core/shell nanoarrays with a holey shell and exposed interfaces for overall water splitting[J]. Journal of Materials Chemistry A, 2019, 7(3): 1196-1205.
[本文引用: 3]
[47]
WANG P C, WAN L, LIN Y Q, et al. MoS2 supported CoS2 on carbon cloth as a high-performance electrode for hydrogen evolution reaction[J]. International Journal of Hydrogen Energy, 2019, 44(31): 16566-16574.
[本文引用: 1]
[48]
ZHANG J, WANG T, LIU P, et al. Engineering water dissociation sites in MoS2 nanosheets for accelerated electrocatalytic hydrogen production[J]. Energy & Environmental Science, 2016, 9(9): 2789-2793.
[本文引用: 1]
[49]
ZOU X X, GOSWAMI A, ASEFA T. Efficient noble metal-free (electro)catalysis of water and alcohol oxidations by zinc-cobalt layered double hydroxide[J]. Journal of the American Chemical Society, 2013, 135(46): 17242-17245.
[本文引用: 1]
[50]
YANG R, ZHOU Y M, XING Y Y, et al. Synergistic coupling of CoFe-LDH arrays with NiFe-LDH nanosheet for highly efficient overall water splitting in alkaline media[J]. Applied Catalysis B: Environmental, 2019, 253: 131-139.
[本文引用: 1]
[51]
YANG Y, YAO H Q, YU Z H, et al. Hierarchical nanoassembly of MoS2 /Co9 S8 /Ni3 S2 /Ni as a highly efficient electrocatalyst for overall water splitting in a wide pH range[J]. Journal of the American Chemical Society, 2019, 141(26): 10417-10430.
[本文引用: 1]
[52]
XU W W, LU Z Y, SUN X M, et al. Superwetting electrodes for gas-involving electrocatalysis[J]. Accounts of Chemical Research, 2018, 51(7): 1590-1598.
[本文引用: 1]
[53]
WANG P C, WAN L, LIN Y Q, et al. NiFe hydroxide supported on hierarchically porous nickel mesh as a high-performance bifunctional electrocatalyst for water splitting at large current density[J]. ChemSusChem, 2019, 12(17): 4038-4045.
[本文引用: 3]
[54]
LI J, ZHU Y Y, CHEN W, et al. Breathing-mimicking electrocatalysis for oxygen evolution and reduction[J]. Joule, 2019, 3(2): 557-569.
[本文引用: 3]
[55]
WANG P C, LIN Y Q, XU Q, et al. Acid-corrosion-induced hollow-structured NiFe-layered double hydroxide electrocatalysts for efficient water oxidation[J]. ACS Applied Energy Materials, 2021, 4(9): 9022-9031.
[本文引用: 4]
[56]
GAO X Q, CHEN Y D, SUN T, et al. Karst landform-featured monolithic electrode for water electrolysis in neutral media[J]. Energy & Environmental Science, 2020, 13(1): 174-182.
[本文引用: 1]
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
4
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
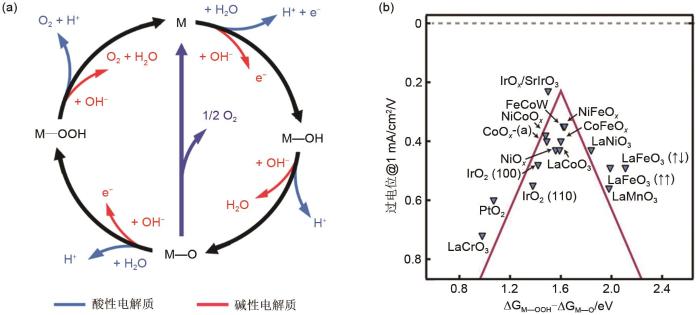
... 与HER过程相比,OER过程更加复杂和缓慢.一般认为碱性条件下OER过程为四电子反应,反应涉及多个反应中间体,如M—OH、M—O和M—OOH(M为吸附位点)
[21 , 27 ] ,这些中间体的形成能存在巨大差异,因此析氧过程的反应动力学更加困难,驱动反应所需要的外界能量也更高.其具体的反应途径如
图3 (a)所示
[23 ] .在碱性条件下,OH
- 首先在活性位点表面失去1个电子形成M—OH中间体.而后在质子和电子转移后,M—OH转化为M—O.紧接着,M—O结合1个OH
- 并失去1个电子转化为M—OOH.最后,M—OOH与另1个氢氧根离子结合同时失去1个电子生成O
2 并暴露出初始活性位点(M).上述析氧过程可用式(6)~
式(9) 表示
图3 <strong>(a)</strong> 氧析出反应途径<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>;<strong>(b)</strong> 氧析出反应火山曲线<sup>[<xref ref-type="bibr" rid="R2">2</xref>]</sup> <strong>(a) The reaction mechanism of OER</strong><sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>;<strong>(b) The volcano plot of OER</strong><sup>[<xref ref-type="bibr" rid="R2">2</xref>]</sup> Fig. 3 ![]()
M + O H - ̿ M ― O H + e - (6) M ― O H + O H - ̿ M ― O + H 2 O + e - (7) M ― O + O H - ̿ M ― O O H + e - (8) M ― O O H + O H - ̿ M + O 2 + H 2 O + e - (9) 如图3 (b)所示,与析氢反应相似,析氧反应也存在“火山”图[2 ] .二氧化铱等贵金属氧化物表现出优异的析氧活性,同时,部分非贵金属氧化物,如氧化镍、氧化钴、二元金属氧化物等也具有高OER催化活性. ...
... [
2 ]
Fig. 3 ![]()
M + O H - ̿ M ― O H + e - (6) M ― O H + O H - ̿ M ― O + H 2 O + e - (7) M ― O + O H - ̿ M ― O O H + e - (8) M ― O O H + O H - ̿ M + O 2 + H 2 O + e - (9) 如图3 (b)所示,与析氢反应相似,析氧反应也存在“火山”图[2 ] .二氧化铱等贵金属氧化物表现出优异的析氧活性,同时,部分非贵金属氧化物,如氧化镍、氧化钴、二元金属氧化物等也具有高OER催化活性. ...
... 如图3 (b)所示,与析氢反应相似,析氧反应也存在“火山”图[2 ] .二氧化铱等贵金属氧化物表现出优异的析氧活性,同时,部分非贵金属氧化物,如氧化镍、氧化钴、二元金属氧化物等也具有高OER催化活性. ...
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
1
... 能源是推动社会进步与变革的强大动力.目前,全球能源供应以化石燃料为主,占比高达80%以上[1 -2 ] ,发展太阳能、风能等可再生能源以推动能源结构快速转型,实现绿色可持续发展是国家重要战略[3 -4 ] .由于太阳能、风能等能源的不连续、不稳定特征,难以直接并入电网,导致可再生能源有效利用率不高[5 ] .在诸多电化学储能技术中,电解水制氢有望大规模消纳可再生能源[6 -8 ] .在我国近期颁布的《氢能产业发展中长期规划(2021—2035年)》中,明确氢能作为战略性新兴产业的定位,是未来国家能源体系的重要组成部分,是终端实现绿色低碳转型的重要载体. ...
1
... 氢气具有诸多的优势:具有高燃烧热值(143 MJ/kg),是同质量焦炭、汽油等化石燃料热值的2~4倍[9 -10 ] .目前主要依靠化石燃料重整制氢,如甲烷重整、煤气化等,具有成本低、原料丰富的优势.化石燃料重整制氢每公斤的成本在1.3~1.5美元之间,而使用电解水制氢的成本则在4美元以上[11 ] ,导致电解水制氢技术在大多数工业过程中缺乏竞争力.世界上超过95%的氢气来源于化石燃料重整制氢,而仅仅只有4%是通过电解水生产[12 ] .随着太阳能发电、风能发电等可再生能源的发展、电力价格不断下降及电解水制氢技术进步带来的能耗减小,可再生能源驱动的电解水制氢成本有望实现与化石燃料重整制氢相当,从而具有市场竞争力.根据中国氢能联盟预测,在2050年可再生能源电解水制氢,将达到总产氢量的70%[13 ] .因此,发展大规模、高效率、长寿命的电解水制氢装备,对支撑氢能产业发展具有重要战略价值. ...
1
... 氢气具有诸多的优势:具有高燃烧热值(143 MJ/kg),是同质量焦炭、汽油等化石燃料热值的2~4倍[9 -10 ] .目前主要依靠化石燃料重整制氢,如甲烷重整、煤气化等,具有成本低、原料丰富的优势.化石燃料重整制氢每公斤的成本在1.3~1.5美元之间,而使用电解水制氢的成本则在4美元以上[11 ] ,导致电解水制氢技术在大多数工业过程中缺乏竞争力.世界上超过95%的氢气来源于化石燃料重整制氢,而仅仅只有4%是通过电解水生产[12 ] .随着太阳能发电、风能发电等可再生能源的发展、电力价格不断下降及电解水制氢技术进步带来的能耗减小,可再生能源驱动的电解水制氢成本有望实现与化石燃料重整制氢相当,从而具有市场竞争力.根据中国氢能联盟预测,在2050年可再生能源电解水制氢,将达到总产氢量的70%[13 ] .因此,发展大规模、高效率、长寿命的电解水制氢装备,对支撑氢能产业发展具有重要战略价值. ...
1
... 氢气具有诸多的优势:具有高燃烧热值(143 MJ/kg),是同质量焦炭、汽油等化石燃料热值的2~4倍[9 -10 ] .目前主要依靠化石燃料重整制氢,如甲烷重整、煤气化等,具有成本低、原料丰富的优势.化石燃料重整制氢每公斤的成本在1.3~1.5美元之间,而使用电解水制氢的成本则在4美元以上[11 ] ,导致电解水制氢技术在大多数工业过程中缺乏竞争力.世界上超过95%的氢气来源于化石燃料重整制氢,而仅仅只有4%是通过电解水生产[12 ] .随着太阳能发电、风能发电等可再生能源的发展、电力价格不断下降及电解水制氢技术进步带来的能耗减小,可再生能源驱动的电解水制氢成本有望实现与化石燃料重整制氢相当,从而具有市场竞争力.根据中国氢能联盟预测,在2050年可再生能源电解水制氢,将达到总产氢量的70%[13 ] .因此,发展大规模、高效率、长寿命的电解水制氢装备,对支撑氢能产业发展具有重要战略价值. ...
1
... 氢气具有诸多的优势:具有高燃烧热值(143 MJ/kg),是同质量焦炭、汽油等化石燃料热值的2~4倍[9 -10 ] .目前主要依靠化石燃料重整制氢,如甲烷重整、煤气化等,具有成本低、原料丰富的优势.化石燃料重整制氢每公斤的成本在1.3~1.5美元之间,而使用电解水制氢的成本则在4美元以上[11 ] ,导致电解水制氢技术在大多数工业过程中缺乏竞争力.世界上超过95%的氢气来源于化石燃料重整制氢,而仅仅只有4%是通过电解水生产[12 ] .随着太阳能发电、风能发电等可再生能源的发展、电力价格不断下降及电解水制氢技术进步带来的能耗减小,可再生能源驱动的电解水制氢成本有望实现与化石燃料重整制氢相当,从而具有市场竞争力.根据中国氢能联盟预测,在2050年可再生能源电解水制氢,将达到总产氢量的70%[13 ] .因此,发展大规模、高效率、长寿命的电解水制氢装备,对支撑氢能产业发展具有重要战略价值. ...
1
... 氢气具有诸多的优势:具有高燃烧热值(143 MJ/kg),是同质量焦炭、汽油等化石燃料热值的2~4倍[9 -10 ] .目前主要依靠化石燃料重整制氢,如甲烷重整、煤气化等,具有成本低、原料丰富的优势.化石燃料重整制氢每公斤的成本在1.3~1.5美元之间,而使用电解水制氢的成本则在4美元以上[11 ] ,导致电解水制氢技术在大多数工业过程中缺乏竞争力.世界上超过95%的氢气来源于化石燃料重整制氢,而仅仅只有4%是通过电解水生产[12 ] .随着太阳能发电、风能发电等可再生能源的发展、电力价格不断下降及电解水制氢技术进步带来的能耗减小,可再生能源驱动的电解水制氢成本有望实现与化石燃料重整制氢相当,从而具有市场竞争力.根据中国氢能联盟预测,在2050年可再生能源电解水制氢,将达到总产氢量的70%[13 ] .因此,发展大规模、高效率、长寿命的电解水制氢装备,对支撑氢能产业发展具有重要战略价值. ...
1
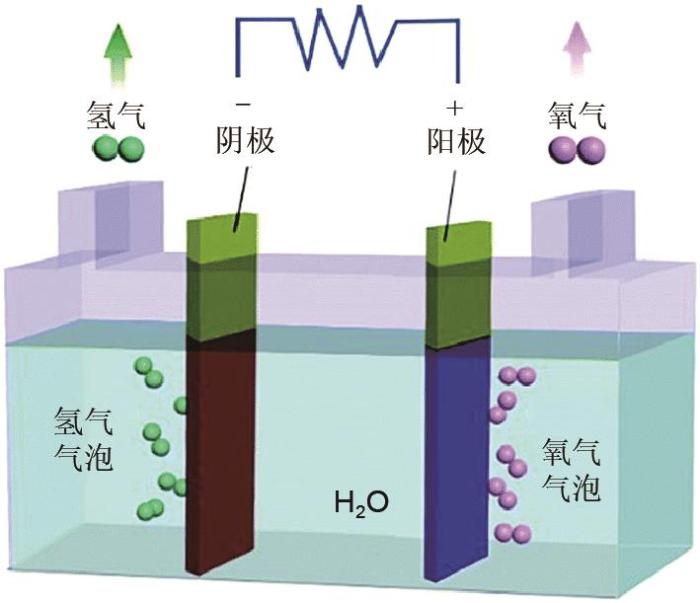
... 电解水制氢技术是一种传统的制氢技术.在1789年电解水现象就被观测到,1800年,Nicholson和Carlisle进一步发展了该技术[14 ] .电解水的原理是通过电能给水提供能量来破坏水分子的氢氧键,从而实现氢气制备.在电极通以一定电流时,阴极发生还原反应析出氢气(析氢反应,HER),阳极发生氧化反应析出氧气(析氧反应,OER),如图1 所示.在碱性条件下,其过程可用如下化学方程式[式(1)~式(3) ]表示 ...
3
... 电解水制氢技术是一种传统的制氢技术.在1789年电解水现象就被观测到,1800年,Nicholson和Carlisle进一步发展了该技术
[14 ] .电解水的原理是通过电能给水提供能量来破坏水分子的氢氧键,从而实现氢气制备.在电极通以一定电流时,阴极发生还原反应析出氢气(析氢反应,HER),阳极发生氧化反应析出氧气(析氧反应,OER),如
图1 所示.在碱性条件下,其过程可用如下化学方程式[式(1)~
式(3) ]表示
H E R : 2 H 2 O + 2 e - ̿ H 2 + 2 O H - (1) O E R : 4 O H - ̿ O 2 + 2 H 2 O + 4 e - (2) 总 反 应 : 2 H 2 O ̿ O 2 + 2 H 2 (3) 图1 电解水电解槽示意图<sup>[<xref ref-type="bibr" rid="R15">15</xref>]</sup> The illustration of the water electrolyzer<sup>[<xref ref-type="bibr" rid="R15">15</xref>]</sup> Fig. 1 ![]()
在酸性条件下,其过程可用如下化学方程式[式(4) 、式(5) ]表示 ...
... [
15 ]
Fig. 1 ![]()
在酸性条件下,其过程可用如下化学方程式[式(4) 、式(5) ]表示 ...
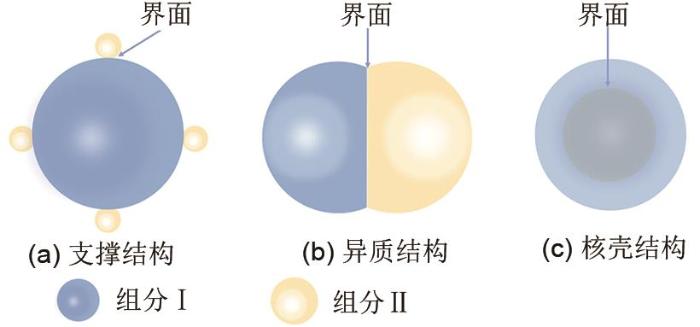
... 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
2
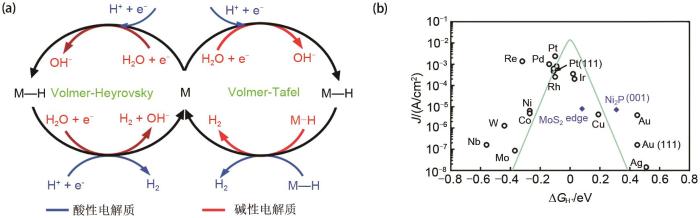
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
... 对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
1
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
1
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
1
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
1
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
7
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
... 与HER过程相比,OER过程更加复杂和缓慢.一般认为碱性条件下OER过程为四电子反应,反应涉及多个反应中间体,如M—OH、M—O和M—OOH(M为吸附位点)[21 , 27 ] ,这些中间体的形成能存在巨大差异,因此析氧过程的反应动力学更加困难,驱动反应所需要的外界能量也更高.其具体的反应途径如图3 (a)所示[23 ] .在碱性条件下,OH- 首先在活性位点表面失去1个电子形成M—OH中间体.而后在质子和电子转移后,M—OH转化为M—O.紧接着,M—O结合1个OH- 并失去1个电子转化为M—OOH.最后,M—OOH与另1个氢氧根离子结合同时失去1个电子生成O2 并暴露出初始活性位点(M).上述析氧过程可用式(6)~式(9) 表示 ...
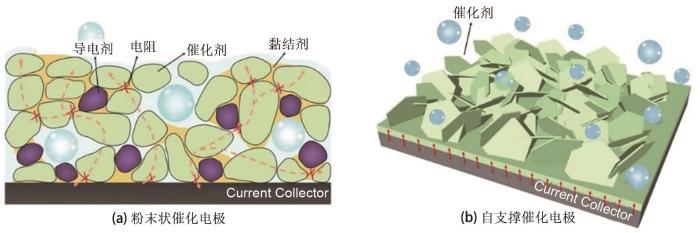
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... [21 ].因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... 对于自支撑催化电极的研究大多数集中在研究开发非贵金属催化材料或超低贵金属负载量的催化材料及其相应的制备方法.不同材料,如过渡金属磷化物、硫化物、碳化物、氮化物、氧化物、氢氧化物、金属合金、MOF等,被广泛研究开发[21 , 29 ] .不同的制备方法,如水热法、溶剂热法、化学气相沉积法、电沉积法等,也被用于制备自支撑催化电极[21 , 33 ] .尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
... [21 , 33 ].尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
... 过去十多年,自支撑催电极受到广泛关注和研究[21 ] .但是,大多数自支撑催化电极的研究主要集中在催化活性物质,很少关注催化基底对催化电极活性与稳定性的影响.本课题组研究过程发现,通过调控基底与催化剂之间的作用力,能够明显提升催化剂-基底界面的稳定性,实现催化电极的长时间使用.通过化学气相沉积法在碳布上负载CoP催化剂,研究碳布基体与CoP催化层之间的固/固界面[37 ] .XPS结果表明碳布氧化处理产生大量的含氧官能团(—C—O、—C=O)将在后续磷化步骤中与CoP催化层产生作用,形成C—O—Co共价键.与之比较,未氧化处理的碳布上负载CoP则不能形成C—O—Co键,两者仅仅通过简单的物理吸附作用结合在一起.另外,研究结果表明,C—O—Co间的化学键,显著提高CoP催化层与碳布基底的结合力,使得CoP催化剂在长时间工作中不易脱落,增加了催化电极的稳定性.因此,通过提升催化电极与基底之间的作用力(成键作用等),增强自支撑催化电极的稳定性,成为高稳定性催化电极的设计原则之一. ...
1
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(Hads ),此后Hads 通过一系列反应形成H2 并从活性位点处脱附[17 ] .在酸性介质中,HER过程的机理如图2 (a)所示.首先H+ 通过Volmer反应(H+ + e- → ads )吸附在阴极上形成Hads 中间体[18 -19 ] .紧接着,Hads 可通过两种反应途径转化为H2 .当Hads 在电极表面活性位点处具有高覆盖率时,Hads 倾向于和另一个相邻的Hads 结合生成H2 ,该过程称为Tafel反应(Hads + Hads → 2 )[20 ] .当Hads 的覆盖率低时,则通过Heyrovsky反应途径(Hads + H+ + e- → 2 )生成H2 [21 ] ,即Hads 同时与1个电子和H+ 结合产生1个H2 .在碱性条件下,由于涉及Hads 形成前需要进行水解离步骤,HER过程更加缓慢[22 ] .HER的反应物也由H+ 变为H2 O,即首先通过Volmer反应(H2 O + e- → ads + OH- )发生解离反应生成吸附的Hads ,而后产生的Hads 通过Heyrovsky或Tafel 反应生成H2 ,其反应过程与酸性条件下类似. ...
5
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附
[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(H
ads ),此后H
ads 通过一系列反应形成H
2 并从活性位点处脱附
[17 ] .在酸性介质中,HER过程的机理如
图2 (a)所示.首先H
+ 通过Volmer反应(H
+ + e
- → H
ads )吸附在阴极上形成H
ads 中间体
[18 -19 ] .紧接着,H
ads 可通过两种反应途径转化为H
2 .当H
ads 在电极表面活性位点处具有高覆盖率时,H
ads 倾向于和另一个相邻的H
ads 结合生成H
2 ,该过程称为Tafel反应(H
ads + H
ads → H
2 )
[20 ] .当H
ads 的覆盖率低时,则通过Heyrovsky反应途径(H
ads + H
+ + e
- → H
2 )生成H
2 [21 ] ,即H
ads 同时与1个电子和H
+ 结合产生1个H
2 .在碱性条件下,由于涉及H
ads 形成前需要进行水解离步骤,HER过程更加缓慢
[22 ] .HER的反应物也由H
+ 变为H
2 O,即首先通过Volmer反应(H
2 O + e
- → H
ads + OH
- )发生解离反应生成吸附的H
ads ,而后产生的H
ads 通过Heyrovsky或Tafel 反应生成H
2 ,其反应过程与酸性条件下类似.
图2 <strong>(a) HER</strong>反应原理图<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>;<strong>(b) HER</strong>火山曲线<sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup> (a) The reaction mechanism of HER<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>; (b) The volcano plot of HER<sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup> Fig. 2 ![]()
对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
... [
23 ]; (b) The volcano plot of HER
[24 ] Fig. 2 ![]()
对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
... 与HER过程相比,OER过程更加复杂和缓慢.一般认为碱性条件下OER过程为四电子反应,反应涉及多个反应中间体,如M—OH、M—O和M—OOH(M为吸附位点)[21 , 27 ] ,这些中间体的形成能存在巨大差异,因此析氧过程的反应动力学更加困难,驱动反应所需要的外界能量也更高.其具体的反应途径如图3 (a)所示[23 ] .在碱性条件下,OH- 首先在活性位点表面失去1个电子形成M—OH中间体.而后在质子和电子转移后,M—OH转化为M—O.紧接着,M—O结合1个OH- 并失去1个电子转化为M—OOH.最后,M—OOH与另1个氢氧根离子结合同时失去1个电子生成O2 并暴露出初始活性位点(M).上述析氧过程可用式(6)~式(9) 表示 ...
... [
23 ];
(b) 氧析出反应火山曲线
[2 ] <strong>(a) The reaction mechanism of OER</strong><sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>;<strong>(b) The volcano plot of OER</strong><sup>[<xref ref-type="bibr" rid="R2">2</xref>]</sup> Fig. 3 ![]()
M + O H - ̿ M ― O H + e - (6) M ― O H + O H - ̿ M ― O + H 2 O + e - (7) M ― O + O H - ̿ M ― O O H + e - (8) M ― O O H + O H - ̿ M + O 2 + H 2 O + e - (9) 如图3 (b)所示,与析氢反应相似,析氧反应也存在“火山”图[2 ] .二氧化铱等贵金属氧化物表现出优异的析氧活性,同时,部分非贵金属氧化物,如氧化镍、氧化钴、二元金属氧化物等也具有高OER催化活性. ...
... [
23 ];
(b) The volcano plot of OER [2 ] Fig. 3 ![]()
M + O H - ̿ M ― O H + e - (6) M ― O H + O H - ̿ M ― O + H 2 O + e - (7) M ― O + O H - ̿ M ― O O H + e - (8) M ― O O H + O H - ̿ M + O 2 + H 2 O + e - (9) 如图3 (b)所示,与析氢反应相似,析氧反应也存在“火山”图[2 ] .二氧化铱等贵金属氧化物表现出优异的析氧活性,同时,部分非贵金属氧化物,如氧化镍、氧化钴、二元金属氧化物等也具有高OER催化活性. ...
3
... 析氢反应属于两电子转移过程,主要包括氢吸附和脱附
[16 ] .电解液中的氢离子首先吸附在电极表面,形成催化活性位点的吸附氢(H
ads ),此后H
ads 通过一系列反应形成H
2 并从活性位点处脱附
[17 ] .在酸性介质中,HER过程的机理如
图2 (a)所示.首先H
+ 通过Volmer反应(H
+ + e
- → H
ads )吸附在阴极上形成H
ads 中间体
[18 -19 ] .紧接着,H
ads 可通过两种反应途径转化为H
2 .当H
ads 在电极表面活性位点处具有高覆盖率时,H
ads 倾向于和另一个相邻的H
ads 结合生成H
2 ,该过程称为Tafel反应(H
ads + H
ads → H
2 )
[20 ] .当H
ads 的覆盖率低时,则通过Heyrovsky反应途径(H
ads + H
+ + e
- → H
2 )生成H
2 [21 ] ,即H
ads 同时与1个电子和H
+ 结合产生1个H
2 .在碱性条件下,由于涉及H
ads 形成前需要进行水解离步骤,HER过程更加缓慢
[22 ] .HER的反应物也由H
+ 变为H
2 O,即首先通过Volmer反应(H
2 O + e
- → H
ads + OH
- )发生解离反应生成吸附的H
ads ,而后产生的H
ads 通过Heyrovsky或Tafel 反应生成H
2 ,其反应过程与酸性条件下类似.
图2 <strong>(a) HER</strong>反应原理图<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>;<strong>(b) HER</strong>火山曲线<sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup> (a) The reaction mechanism of HER<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>; (b) The volcano plot of HER<sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup> Fig. 2 ![]()
对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
... [
24 ]
Fig. 2 ![]()
对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
... 对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
1
... 对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
2
... 对于不同的HER催化剂,其反应机理不同.一般可以通过塔菲尔斜率来判断决速步骤,从而推断出反应途径.当塔菲尔斜率约为120 mV/dec时,Volmer步骤为决速步骤;当塔菲尔斜率约为40 mV/dec时,Heyrovsky步骤为决速步骤;当塔菲尔斜率约为30 mV/dec时,Tafel步骤为决速步骤[16 , 25 ] .在HER过程中,通过密度泛函理论(DFT)计算得到的氢吸附自由能(∆G H )是评价电催化剂性能的重要参数.较负的∆G H 值表明氢在电极上的吸附比脱附容易,因此Heyrovsky或者Tafel 反应是决速步骤[26 ] .反之,较正的∆G H 值代表催化剂和Hads 的相互作用较弱,决速步骤是Volmer反应[26 ] .因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
... [26 ].因此,高催化活性的HER电催化剂应具有接近于零的∆G H ,从而实现氢吸附和脱附的平衡.为此,人们将不同催化剂的催化活性与∆G H 的关系做图得到“火山”曲线[图2 (b)][24 ] .从中可以得到贵金属Pt具有优异的HER活性,此外多种非贵金属化合物也表现出高HER活性,如二硫化钼和磷化镍等. ...
1
... 与HER过程相比,OER过程更加复杂和缓慢.一般认为碱性条件下OER过程为四电子反应,反应涉及多个反应中间体,如M—OH、M—O和M—OOH(M为吸附位点)[21 , 27 ] ,这些中间体的形成能存在巨大差异,因此析氧过程的反应动力学更加困难,驱动反应所需要的外界能量也更高.其具体的反应途径如图3 (a)所示[23 ] .在碱性条件下,OH- 首先在活性位点表面失去1个电子形成M—OH中间体.而后在质子和电子转移后,M—OH转化为M—O.紧接着,M—O结合1个OH- 并失去1个电子转化为M—OOH.最后,M—OOH与另1个氢氧根离子结合同时失去1个电子生成O2 并暴露出初始活性位点(M).上述析氧过程可用式(6)~式(9) 表示 ...
2
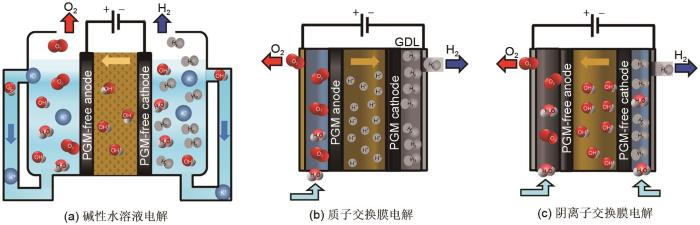
... 电解槽是电解水制氢的关键部分.迄今为止,主要有3种不同类型的低温电解水制氢技术(
图4 ),即碱性水溶液电解水、质子交换膜电解水和阴离子交换膜电解水.目前发展最成熟的电解水制氢工艺是碱性水溶液电解水技术,具有操作简便、成本低廉的优势,已实现大规模商业化运用.但是,该过程存在电解水效率低、响应速度慢、高碱浓度对电解水设备要求严苛的缺点.为了提供碱性水溶液电解水技术的效率,需要重点关注电解槽结构设计并高效催化电极.质子交换膜电解水制氢技术则能够有效解决这些问题——通过使用致密隔膜可以实现迅速动态响应;电极与隔膜的零间距接触,能够显著提升电解效率;电解过程使用纯水,可以解决碱液对设备腐蚀问题.但是质子交换膜电解水技术使用昂贵的贵金属催化剂,导致了制氢的高成本.为此,亟需开发低成本、高活性的替代催化剂.阴离子交换膜电解水则结合了碱性水溶液电解水技术的低成本优势和质子交换膜电解水的高效率优势,有望成为大规模电能转化为氢能的储能技术.但是目前同样缺乏在碱性条件下具有高稳定的催化剂.
图4 <strong>3</strong>种低温电解水制氢技术示意图:<strong>(a)</strong> 碱性水溶液电解水技术;<strong>(b)</strong> 质子交换膜电解水技术;<strong>(c)</strong> 阴离子交换膜电解水技术<sup>[<xref ref-type="bibr" rid="R28">28</xref>]</sup> The schmatic diagrams of three typical water-splitting electrolyzers: (a) Alkaline water electrolyzer;(b) Proton exchange membrane water electrolyzers; (c) Alkaline exchange membrane water electrolyzers<sup>[<xref ref-type="bibr" rid="R28">28</xref>]</sup> Fig. 4 ![]()
综上所述,三种电解水制氢技术的进一步发展都极度依赖高性能催化剂.为此,研究开发高活性、高稳定性、低成本的催化电极材料是推动电解水制氢技术发展的关键. ...
... [
28 ]
Fig. 4 ![]()
综上所述,三种电解水制氢技术的进一步发展都极度依赖高性能催化剂.为此,研究开发高活性、高稳定性、低成本的催化电极材料是推动电解水制氢技术发展的关键. ...
2
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... 对于自支撑催化电极的研究大多数集中在研究开发非贵金属催化材料或超低贵金属负载量的催化材料及其相应的制备方法.不同材料,如过渡金属磷化物、硫化物、碳化物、氮化物、氧化物、氢氧化物、金属合金、MOF等,被广泛研究开发[21 , 29 ] .不同的制备方法,如水热法、溶剂热法、化学气相沉积法、电沉积法等,也被用于制备自支撑催化电极[21 , 33 ] .尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
1
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
1
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
2
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... 基于对3个关键的认识,课题组发展了自源生长方法(将基底同时作为反应物与导电材料),采用“酸刻蚀/气相生长”法制备自支撑催化析氧电极的绿色工艺方法.将基底元素直接作为反应物可以避免使用外加物质,此时原位生长的催化剂与基底将会有更高的兼容性,催化剂与基底能够实现“一体化”结合.通过酸刻蚀法对泡沫镍铁表面进行原位刻蚀(常温、常压),使用空气中的氧气进行氧化反应,在泡沫镍铁表面原位生长NiFe LDH纳米片阵列[图12 (a)],即自支撑镍铁@镍铁阳极(NiFe LDH@NiFe)[55 ] .NiFe LDH@NiFe催化电极表现出了优异的催化活性,在电流密度为10 mA/cm2 时所需的析氧过电位仅仅为201 mV,远优于大多数文献[55 ] 报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...
2
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... 对于自支撑催化电极的研究大多数集中在研究开发非贵金属催化材料或超低贵金属负载量的催化材料及其相应的制备方法.不同材料,如过渡金属磷化物、硫化物、碳化物、氮化物、氧化物、氢氧化物、金属合金、MOF等,被广泛研究开发[21 , 29 ] .不同的制备方法,如水热法、溶剂热法、化学气相沉积法、电沉积法等,也被用于制备自支撑催化电极[21 , 33 ] .尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
1
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
3
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展. ...
... 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
... Zhang等
[43 ] 报道了一种Ni(OH)
2 与MoS
2 复合的催化电极(Ni(OH)
2 /MoS
2 )用于催化析氢[
图9 (a)].其中Ni(OH)
2 纳米颗粒(5~10 nm)均匀地负载在MoS
2 纳米片表面形成支撑结构复合物.透射电子显微镜(TEM)结果表明在MoS
2 纳米片表面引入Ni(OH)
2 纳米粒子将增大MoS
2 的层间距,从而暴露出更多硫活性边缘.X射线光电子能谱(XPS)测试结果表明引入Ni(OH)
2 后,Ni(OH)
2 /MoS
2 中S 2p的结合能负移,说明电子从Ni(OH)
2 转移到MoS
2 ,证明两组分之间存在强电子作用.DFT结果表明在两组分的界面处,Ni(OH)
2 易于和O结合而MoS
2 易于与H结合[
图9 (b)、(c)].因此,水分子首先吸附在Ni(OH)
2 上并发生解离,同时解离的氢被吸附在临近的MoS
2 活性位点,最后两个吸附态氢结合生成H
2 .因此,MoS
2 与Ni(OH)
2 的协同作用能显著加快HER动力学.另外,Niu等人
[44 ] 通过两步电沉积在泡沫镍上原位生长多级结构的FeOOH/Ni(OH)
2 ,Ni与Fe之间的协同作用使得FeOOH/Ni(OH)
2 具有高催化活性,电流密度为40 mA/cm
2 时所需析氧过电位仅为207 mV.
图8 三种典型的两组分化合物组合的复合物结构示意图 <strong>(a)</strong> 支撑结构;<strong>(b)</strong> 异质结构;<strong>(c)</strong> 核壳结构<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup> Schematic diagram of three typical components with two compositions: (a) Supported structure; (b) Heterostructure; (c) Core-shell structure Fig. 8 ![]()
图9 <strong>(a) MoS<sub>2</sub>/NiCo-LDH</strong>复合材料合成示意图;<strong>(b)</strong>~<strong>(c) MoS<sub>2</sub></strong> 边缘和<strong>Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 的<strong>HER</strong>自由能<sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>;<strong>(d) Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 合成示意图<sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>;<strong>(e) Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub></strong> 的合成示意图<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> (a) The schematic diagram of fabrication of MoS<sub>2</sub>/NiCo-LDH; (b)-(c) The HER free energy of the edge of MoS<sub>2</sub> and interface of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>; (d) Fabrication of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>; (e) Fabrication of Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub><sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
2
... 目前,催化电极的类型可分为两类:粉末状催化电极与自支撑催化电极.其中,粉末状催化电极是将粉末催化剂使用黏结剂固定在导电基底上,制备形成催化电极[
图5 (a)];自支撑催化电极是将催化活性物质原位生长在导电基底上,直接得到“一体化”三维催化电极,避免了黏结剂的使用[
图5 (b)].由于电催化剂制备技术研究历史和主要应用领域的原因,大多数制备的是粉末状催化电极.但是,它在电解水制氢过程中往往存在以下几方面问题:①使用亲水性差的黏结剂,不可避免增加传质阻力,并会覆盖催化活性位点
[21 , 29 ] ;②粉末催化剂与导电基底间的黏附力低,导致低负载量(通常小于1 mg/cm
2 ),从而使提供的催化活性位点有限;③长时间大电流密度电解过程中,粉末催化剂容易从导电基底上脱落,引起电催化性能急剧下降
[30 ] .为此,与传统的粉末状催化电极相比,自支撑催化电极具有以下优势:①催化剂在导电基底的原位生长能避免黏结剂的使用,简化电极制备过程,降低制备成本
[31 ] ;②导电基底可分散催化剂并提高催化剂负载量,从而提供丰富的催化活性位点
[32 ] ;③能确保催化层与导电基底的结合紧密,确保电荷的快速转移并防止催化剂脱落
[33 -34 ] ;④通过对电极表面形貌和微观结构的合理调控,自支撑催化电极更容易实现表面亲水/疏气工程,使得电解液完全浸润活性位点并加快气泡产物的脱附,增强电催化剂与电解液接触,促进电荷和离子转移
[35 ] .这些优点有利于自支撑催化电极在大电流密度条件下具有优异的催化活性并保持长期电解稳定性
[21 ] .因此,近几年,自支撑催化电极在设计与制备方面取得显著进展.
图5 粉末型催化电极与自支撑催化电极结构比较<sup>[<xref ref-type="bibr" rid="R36">36</xref>]</sup><strong>: (a)</strong> 粉末型催化电极;<strong>(b)</strong> 自支撑催化电极 The comparison of powdery electrode and self-supported electrode<sup>[<xref ref-type="bibr" rid="R36">36</xref>]</sup>: (a) Powdery electrode;(b) Self-supported electrode Fig. 5 ![]()
对于自支撑催化电极的研究大多数集中在研究开发非贵金属催化材料或超低贵金属负载量的催化材料及其相应的制备方法.不同材料,如过渡金属磷化物、硫化物、碳化物、氮化物、氧化物、氢氧化物、金属合金、MOF等,被广泛研究开发[21 , 29 ] .不同的制备方法,如水热法、溶剂热法、化学气相沉积法、电沉积法等,也被用于制备自支撑催化电极[21 , 33 ] .尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
... [
36 ]: (a) Powdery electrode;(b) Self-supported electrode
Fig. 5 ![]()
对于自支撑催化电极的研究大多数集中在研究开发非贵金属催化材料或超低贵金属负载量的催化材料及其相应的制备方法.不同材料,如过渡金属磷化物、硫化物、碳化物、氮化物、氧化物、氢氧化物、金属合金、MOF等,被广泛研究开发[21 , 29 ] .不同的制备方法,如水热法、溶剂热法、化学气相沉积法、电沉积法等,也被用于制备自支撑催化电极[21 , 33 ] .尽管近年来用于电解水的自支撑催化电极取得显著成果,但是其依旧没有实现商业化.这主要是因为在科学合理设计自支撑催化电极时缺乏深刻理解,自支撑催化电极的活性与稳定性还不能满足工业化上万小时的稳定工作.为此,研究开发可商业化自支撑催化电极要加深对自支撑催化电极在多维度上的理解. ...
1
... 过去十多年,自支撑催电极受到广泛关注和研究[21 ] .但是,大多数自支撑催化电极的研究主要集中在催化活性物质,很少关注催化基底对催化电极活性与稳定性的影响.本课题组研究过程发现,通过调控基底与催化剂之间的作用力,能够明显提升催化剂-基底界面的稳定性,实现催化电极的长时间使用.通过化学气相沉积法在碳布上负载CoP催化剂,研究碳布基体与CoP催化层之间的固/固界面[37 ] .XPS结果表明碳布氧化处理产生大量的含氧官能团(—C—O、—C=O)将在后续磷化步骤中与CoP催化层产生作用,形成C—O—Co共价键.与之比较,未氧化处理的碳布上负载CoP则不能形成C—O—Co键,两者仅仅通过简单的物理吸附作用结合在一起.另外,研究结果表明,C—O—Co间的化学键,显著提高CoP催化层与碳布基底的结合力,使得CoP催化剂在长时间工作中不易脱落,增加了催化电极的稳定性.因此,通过提升催化电极与基底之间的作用力(成键作用等),增强自支撑催化电极的稳定性,成为高稳定性催化电极的设计原则之一. ...
3
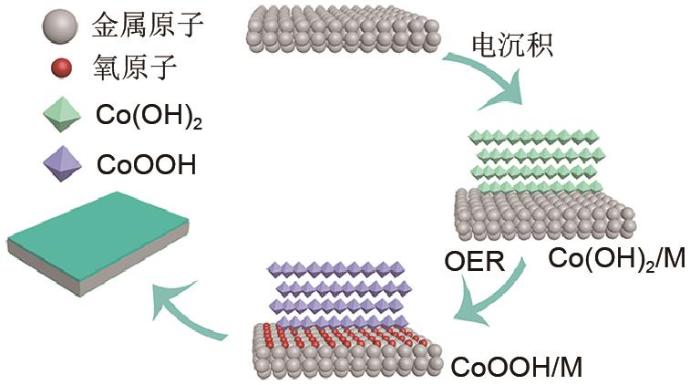
... 为了研究催化基底对催化电极稳定性的影响,通过一步电化学沉积法将Co(OH)2 纳米层沉积在不同的金属箔表面(Fe、Co、Ni、Cu、Ag、Ti),并直接作为OER催化电极(图7 )[38 ] .研究表明催化电极的活性取决于Co(OH)2 负载的基底,其中Fe基底支撑Co(OH)2 催化电极表现出最高OER活性.研究结果阐明在OER过程中金属基底表面会被原位氧化形成氧化层(金属羟基氧化物、氢氧化物、氧化物),同时Co(OH)2 催化层在OER过程会转化为具有高OER催化活性的CoOOH催化组分.原位形成的氧化层与原位转换的CoOOH催化层之间存在强电子相互作用,改变了催化剂-基底界面处的电子结构,从而改变不同金属支撑的Co(OH)2 电极的OER反应动力学,并表现出不同的OER催化活性.因此,该工作阐明通过导电基底与催化层的匹配设计是制备高性能自支撑催化电极的有效方法.通过研究催化剂-基底界面对自支撑催化电极催化活性的影响,为设计高效的催化电极提供新的研究思路和设计原则,不仅要注重提升催化层的催化活性,还要注重提高催化层与催化基底的匹配度. ...
... [
38 ]
The schematic diagram of frabrication of Co(OH)<sub>2</sub> based electrode<sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup> Fig. 7 ![]()
<strong>2.2</strong> 催化剂内部界面 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
... [
38 ]
Fig. 7 ![]()
<strong>2.2</strong> 催化剂内部界面 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
1
... 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
1
... 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
1
... 将单组份结构催化剂发展为多组分结构催化剂是提升催化剂催化活性的一种有效策略,尤其在多相催化领域[39 -41 ] .对于HER和OER,其本征反应速率受到催化剂对反应中间体与反应物的化学吸附作用的影响[15 , 42 ] .通过设计催化剂内部的界面将能够优化反应物或反应中间体的化学吸附作用力,从而加速反应速率.对于金属化合物复合物,最为广泛研究的是两组分复合物.根据两组分的组成结构不同,两组分的复合结构可以分为支撑结构、异质结构和核壳结构三种典型结构模型[图(8)][35 ] .下面就3种关键界面进行详细的介绍. ...
3
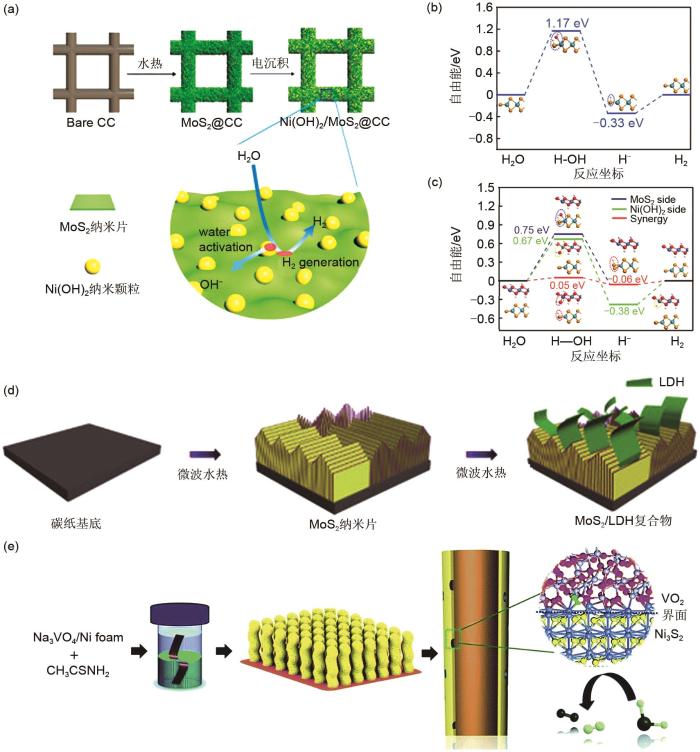
... Zhang等[43 ] 报道了一种Ni(OH)2 与MoS2 复合的催化电极(Ni(OH)2 /MoS2 )用于催化析氢[图9 (a)].其中Ni(OH)2 纳米颗粒(5~10 nm)均匀地负载在MoS2 纳米片表面形成支撑结构复合物.透射电子显微镜(TEM)结果表明在MoS2 纳米片表面引入Ni(OH)2 纳米粒子将增大MoS2 的层间距,从而暴露出更多硫活性边缘.X射线光电子能谱(XPS)测试结果表明引入Ni(OH)2 后,Ni(OH)2 /MoS2 中S 2p的结合能负移,说明电子从Ni(OH)2 转移到MoS2 ,证明两组分之间存在强电子作用.DFT结果表明在两组分的界面处,Ni(OH)2 易于和O结合而MoS2 易于与H结合[图9 (b)、(c)].因此,水分子首先吸附在Ni(OH)2 上并发生解离,同时解离的氢被吸附在临近的MoS2 活性位点,最后两个吸附态氢结合生成H2 .因此,MoS2 与Ni(OH)2 的协同作用能显著加快HER动力学.另外,Niu等人[44 ] 通过两步电沉积在泡沫镍上原位生长多级结构的FeOOH/Ni(OH)2 ,Ni与Fe之间的协同作用使得FeOOH/Ni(OH)2 具有高催化活性,电流密度为40 mA/cm2 时所需析氧过电位仅为207 mV. ...
... [
43 ];
(d) Ni(OH)2 /MoS2 合成示意图
[45 ] ;
(e) Ni3 S2 /VO2 的合成示意图
[46 ] (a) The schematic diagram of fabrication of MoS<sub>2</sub>/NiCo-LDH; (b)-(c) The HER free energy of the edge of MoS<sub>2</sub> and interface of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>; (d) Fabrication of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>; (e) Fabrication of Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub><sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
... [
43 ]; (d) Fabrication of Ni(OH)
2 /MoS
2 [45 ] ; (e) Fabrication of Ni
3 S
2 /VO
2 [46 ] Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
1
... Zhang等[43 ] 报道了一种Ni(OH)2 与MoS2 复合的催化电极(Ni(OH)2 /MoS2 )用于催化析氢[图9 (a)].其中Ni(OH)2 纳米颗粒(5~10 nm)均匀地负载在MoS2 纳米片表面形成支撑结构复合物.透射电子显微镜(TEM)结果表明在MoS2 纳米片表面引入Ni(OH)2 纳米粒子将增大MoS2 的层间距,从而暴露出更多硫活性边缘.X射线光电子能谱(XPS)测试结果表明引入Ni(OH)2 后,Ni(OH)2 /MoS2 中S 2p的结合能负移,说明电子从Ni(OH)2 转移到MoS2 ,证明两组分之间存在强电子作用.DFT结果表明在两组分的界面处,Ni(OH)2 易于和O结合而MoS2 易于与H结合[图9 (b)、(c)].因此,水分子首先吸附在Ni(OH)2 上并发生解离,同时解离的氢被吸附在临近的MoS2 活性位点,最后两个吸附态氢结合生成H2 .因此,MoS2 与Ni(OH)2 的协同作用能显著加快HER动力学.另外,Niu等人[44 ] 通过两步电沉积在泡沫镍上原位生长多级结构的FeOOH/Ni(OH)2 ,Ni与Fe之间的协同作用使得FeOOH/Ni(OH)2 具有高催化活性,电流密度为40 mA/cm2 时所需析氧过电位仅为207 mV. ...
3
... Zhang等
[43 ] 报道了一种Ni(OH)
2 与MoS
2 复合的催化电极(Ni(OH)
2 /MoS
2 )用于催化析氢[
图9 (a)].其中Ni(OH)
2 纳米颗粒(5~10 nm)均匀地负载在MoS
2 纳米片表面形成支撑结构复合物.透射电子显微镜(TEM)结果表明在MoS
2 纳米片表面引入Ni(OH)
2 纳米粒子将增大MoS
2 的层间距,从而暴露出更多硫活性边缘.X射线光电子能谱(XPS)测试结果表明引入Ni(OH)
2 后,Ni(OH)
2 /MoS
2 中S 2p的结合能负移,说明电子从Ni(OH)
2 转移到MoS
2 ,证明两组分之间存在强电子作用.DFT结果表明在两组分的界面处,Ni(OH)
2 易于和O结合而MoS
2 易于与H结合[
图9 (b)、(c)].因此,水分子首先吸附在Ni(OH)
2 上并发生解离,同时解离的氢被吸附在临近的MoS
2 活性位点,最后两个吸附态氢结合生成H
2 .因此,MoS
2 与Ni(OH)
2 的协同作用能显著加快HER动力学.另外,Niu等人
[44 ] 通过两步电沉积在泡沫镍上原位生长多级结构的FeOOH/Ni(OH)
2 ,Ni与Fe之间的协同作用使得FeOOH/Ni(OH)
2 具有高催化活性,电流密度为40 mA/cm
2 时所需析氧过电位仅为207 mV.
图8 三种典型的两组分化合物组合的复合物结构示意图 <strong>(a)</strong> 支撑结构;<strong>(b)</strong> 异质结构;<strong>(c)</strong> 核壳结构<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup> Schematic diagram of three typical components with two compositions: (a) Supported structure; (b) Heterostructure; (c) Core-shell structure Fig. 8 ![]()
图9 <strong>(a) MoS<sub>2</sub>/NiCo-LDH</strong>复合材料合成示意图;<strong>(b)</strong>~<strong>(c) MoS<sub>2</sub></strong> 边缘和<strong>Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 的<strong>HER</strong>自由能<sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>;<strong>(d) Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 合成示意图<sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>;<strong>(e) Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub></strong> 的合成示意图<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> (a) The schematic diagram of fabrication of MoS<sub>2</sub>/NiCo-LDH; (b)-(c) The HER free energy of the edge of MoS<sub>2</sub> and interface of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>; (d) Fabrication of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>; (e) Fabrication of Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub><sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
... [
45 ]; (e) Fabrication of Ni
3 S
2 /VO
2 [46 ] Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
... 两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
3
... Zhang等
[43 ] 报道了一种Ni(OH)
2 与MoS
2 复合的催化电极(Ni(OH)
2 /MoS
2 )用于催化析氢[
图9 (a)].其中Ni(OH)
2 纳米颗粒(5~10 nm)均匀地负载在MoS
2 纳米片表面形成支撑结构复合物.透射电子显微镜(TEM)结果表明在MoS
2 纳米片表面引入Ni(OH)
2 纳米粒子将增大MoS
2 的层间距,从而暴露出更多硫活性边缘.X射线光电子能谱(XPS)测试结果表明引入Ni(OH)
2 后,Ni(OH)
2 /MoS
2 中S 2p的结合能负移,说明电子从Ni(OH)
2 转移到MoS
2 ,证明两组分之间存在强电子作用.DFT结果表明在两组分的界面处,Ni(OH)
2 易于和O结合而MoS
2 易于与H结合[
图9 (b)、(c)].因此,水分子首先吸附在Ni(OH)
2 上并发生解离,同时解离的氢被吸附在临近的MoS
2 活性位点,最后两个吸附态氢结合生成H
2 .因此,MoS
2 与Ni(OH)
2 的协同作用能显著加快HER动力学.另外,Niu等人
[44 ] 通过两步电沉积在泡沫镍上原位生长多级结构的FeOOH/Ni(OH)
2 ,Ni与Fe之间的协同作用使得FeOOH/Ni(OH)
2 具有高催化活性,电流密度为40 mA/cm
2 时所需析氧过电位仅为207 mV.
图8 三种典型的两组分化合物组合的复合物结构示意图 <strong>(a)</strong> 支撑结构;<strong>(b)</strong> 异质结构;<strong>(c)</strong> 核壳结构<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup> Schematic diagram of three typical components with two compositions: (a) Supported structure; (b) Heterostructure; (c) Core-shell structure Fig. 8 ![]()
图9 <strong>(a) MoS<sub>2</sub>/NiCo-LDH</strong>复合材料合成示意图;<strong>(b)</strong>~<strong>(c) MoS<sub>2</sub></strong> 边缘和<strong>Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 的<strong>HER</strong>自由能<sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>;<strong>(d) Ni(OH)<sub>2</sub>/MoS<sub>2</sub></strong> 合成示意图<sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>;<strong>(e) Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub></strong> 的合成示意图<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> (a) The schematic diagram of fabrication of MoS<sub>2</sub>/NiCo-LDH; (b)-(c) The HER free energy of the edge of MoS<sub>2</sub> and interface of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>; (d) Fabrication of Ni(OH)<sub>2</sub>/MoS<sub>2</sub><sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>; (e) Fabrication of Ni<sub>3</sub>S<sub>2</sub>/VO<sub>2</sub><sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
... [
46 ]
Fig. 9 ![]()
两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
... 两组分核壳结构的复合催化电极也被广泛研究.Lyu等[46 ] 通过一步水热法在泡沫镍上合成核壳结构的Ni3 S2 /VO2 纳米线阵列[图9 (e)].XPS结果表明Ni3 S2 /VO2 中Ni 2p键能相比单组分Ni3 S2 的Ni 2p键能出现明显正偏移,表明Ni3 S2 与VO2 之间存在强电子相互作用,将使得界面的电子密度重新分布.另外,DFT结果证明Ni3 S2 与VO2 的界面能够显著降低OER的吉布斯自由能和减小d带中心,从而显著加快OER和HER的反应速度.另外,也可以通过耦合两种层状双金属氢氧化物来设计高性能催化电极.Yang等[50 ] 通过两步法在泡沫镍表面原位生长NiFe LDH修饰的CoFe LDH纳米片阵列(CoFe@NiFe/NF).SEM测试结果证实NiFe LDH纳米片垂直沉积在CoFe LDH纳米片上.这种独特的结构一方面提供了更大的比表面积,另一方面提供了传质通道.电化学数据表明CoFe@NiFe/NF同时具有高HER和OER催化性能.将它们同时作为阳极与阴极,CoFe@NiFe/NF电极装配的电解槽仅需1.59 V就可达到10 mA/cm2 .此外,三元组分的核壳结构的复合物也被研究.Yang等[51 ] 通过一步水热法合成了一种核壳结构的MoS2 /Co9 S8 /Ni3 S2 纳米棒阵列用于全电解水,其中Ni3 S2 为纳米棒结构,而MoS2 纳米片和Co9 S8 纳米片则均匀地负载在Ni3 S2 上.研究表明,MoS2 和Co9 S8 之间的协同作用可以增强电子转移速度,同时Ni3 S2 作为电子转移通道有利于电子的快速传输.为此,MoS2 /Co9 S8 /Ni3 S2 催化电极具有高催化性能,使用MoS2 /Co9 S8 /Ni3 S2 作为阳极和阴极用于全电解水,在10 mA/cm2 时所需的电解电压仅仅为1.54 V,优于大多数研究报道. ...
1
... 两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
1
... 两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
1
... 两组分异质结构的复合催化电极得到深入研究.本课题通过结合电沉积法和水热法制备了一种异质结构的MoS2 /CoS2 复合催化电极[47 ] .研究表明MoS2 /CoS2 直接的界面将会显著增加硫活性边缘位点,从而显著增强MoS2 /CoS催化电极在酸性条件下的催化析氢活性,在电流密度为10 mA/cm2 时所需析氢过电位仅仅为118 mV,优于大多数MoS2 基析氢催化剂.尽管MoS2 在酸性介质中具有高催化析氢活性,其在碱性环境下由于羟基不被MoS2 吸附,MoS2 在碱性下的析氢动力学十分缓慢[48 ] .层状双金属氢氧化物(LDH)在碱性条件下表现高析氧活性,能够有效地吸附羟基从而促进水的解离[49 ] .为此,通过结合MoS2 与LDH可以增强MoS2 在碱性条件下的析氢性能.Hu等[45 ] 通过两步微波水热法合成具有异质结构的MoS2 /NiCo LDH复合催化电极[图9 (d)].TEM测试结果证明MoS2 与NiCo LDH两组分存在大量异质界面,由MoS2 的(022)面与NiCo LDH的(015)面构成.拉曼光谱测试结果分析证明MoS2 /NiCo LDH中MoS2 的A 1g 模显著减小,而E 2g 1 显著增加,表明MoS2 与NiCo LDH之间存在强电子相互作用,能提升MoS2 /NiCo LDH的催化活性. ...
1
... 两组分核壳结构的复合催化电极也被广泛研究.Lyu等[46 ] 通过一步水热法在泡沫镍上合成核壳结构的Ni3 S2 /VO2 纳米线阵列[图9 (e)].XPS结果表明Ni3 S2 /VO2 中Ni 2p键能相比单组分Ni3 S2 的Ni 2p键能出现明显正偏移,表明Ni3 S2 与VO2 之间存在强电子相互作用,将使得界面的电子密度重新分布.另外,DFT结果证明Ni3 S2 与VO2 的界面能够显著降低OER的吉布斯自由能和减小d带中心,从而显著加快OER和HER的反应速度.另外,也可以通过耦合两种层状双金属氢氧化物来设计高性能催化电极.Yang等[50 ] 通过两步法在泡沫镍表面原位生长NiFe LDH修饰的CoFe LDH纳米片阵列(CoFe@NiFe/NF).SEM测试结果证实NiFe LDH纳米片垂直沉积在CoFe LDH纳米片上.这种独特的结构一方面提供了更大的比表面积,另一方面提供了传质通道.电化学数据表明CoFe@NiFe/NF同时具有高HER和OER催化性能.将它们同时作为阳极与阴极,CoFe@NiFe/NF电极装配的电解槽仅需1.59 V就可达到10 mA/cm2 .此外,三元组分的核壳结构的复合物也被研究.Yang等[51 ] 通过一步水热法合成了一种核壳结构的MoS2 /Co9 S8 /Ni3 S2 纳米棒阵列用于全电解水,其中Ni3 S2 为纳米棒结构,而MoS2 纳米片和Co9 S8 纳米片则均匀地负载在Ni3 S2 上.研究表明,MoS2 和Co9 S8 之间的协同作用可以增强电子转移速度,同时Ni3 S2 作为电子转移通道有利于电子的快速传输.为此,MoS2 /Co9 S8 /Ni3 S2 催化电极具有高催化性能,使用MoS2 /Co9 S8 /Ni3 S2 作为阳极和阴极用于全电解水,在10 mA/cm2 时所需的电解电压仅仅为1.54 V,优于大多数研究报道. ...
1
... 两组分核壳结构的复合催化电极也被广泛研究.Lyu等[46 ] 通过一步水热法在泡沫镍上合成核壳结构的Ni3 S2 /VO2 纳米线阵列[图9 (e)].XPS结果表明Ni3 S2 /VO2 中Ni 2p键能相比单组分Ni3 S2 的Ni 2p键能出现明显正偏移,表明Ni3 S2 与VO2 之间存在强电子相互作用,将使得界面的电子密度重新分布.另外,DFT结果证明Ni3 S2 与VO2 的界面能够显著降低OER的吉布斯自由能和减小d带中心,从而显著加快OER和HER的反应速度.另外,也可以通过耦合两种层状双金属氢氧化物来设计高性能催化电极.Yang等[50 ] 通过两步法在泡沫镍表面原位生长NiFe LDH修饰的CoFe LDH纳米片阵列(CoFe@NiFe/NF).SEM测试结果证实NiFe LDH纳米片垂直沉积在CoFe LDH纳米片上.这种独特的结构一方面提供了更大的比表面积,另一方面提供了传质通道.电化学数据表明CoFe@NiFe/NF同时具有高HER和OER催化性能.将它们同时作为阳极与阴极,CoFe@NiFe/NF电极装配的电解槽仅需1.59 V就可达到10 mA/cm2 .此外,三元组分的核壳结构的复合物也被研究.Yang等[51 ] 通过一步水热法合成了一种核壳结构的MoS2 /Co9 S8 /Ni3 S2 纳米棒阵列用于全电解水,其中Ni3 S2 为纳米棒结构,而MoS2 纳米片和Co9 S8 纳米片则均匀地负载在Ni3 S2 上.研究表明,MoS2 和Co9 S8 之间的协同作用可以增强电子转移速度,同时Ni3 S2 作为电子转移通道有利于电子的快速传输.为此,MoS2 /Co9 S8 /Ni3 S2 催化电极具有高催化性能,使用MoS2 /Co9 S8 /Ni3 S2 作为阳极和阴极用于全电解水,在10 mA/cm2 时所需的电解电压仅仅为1.54 V,优于大多数研究报道. ...
1
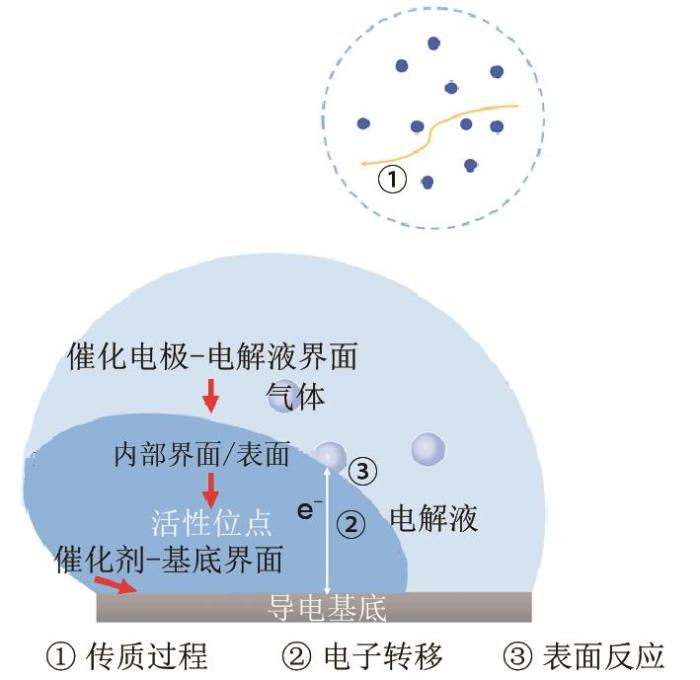
... 在析氢/析氧反应时,催化电极直接与电解液接触,两者的接触状态将会显著影响催化电极的电荷转移速度与传质速度.例如,HER反应时在大电流密度下将会在电极表面产生大量的氢气气泡,这将会严重的阻碍电解液浸润活性位点,导致高反应电阻[52 ] .为此,为加快析氢和析氧反应动力学,需要构建一个理想的电极-电解液界面以促进反应物的传输和反应产物的脱附. ...
3
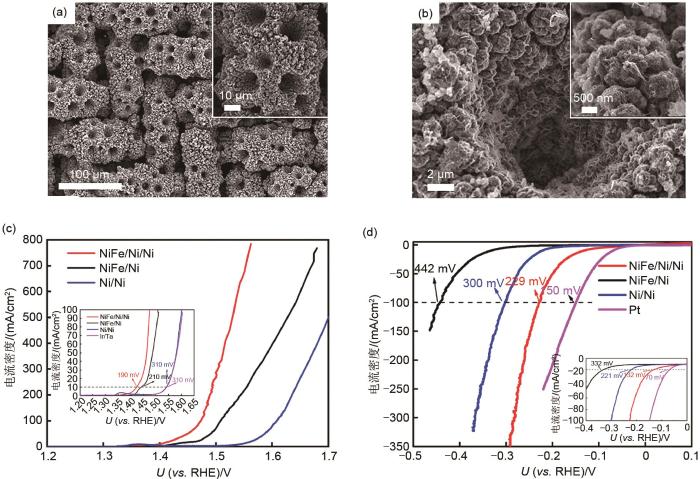
... 为了构建有序传质通道,实现气液固三相传质过程强化,课题组通过两步电沉积法制备得到三维多级结构的NiFe/Ni/Ni催化电极[53 ] .其形貌如图10 所示,图10 (a)表明在镍网上形成了大量有序的传质通道,图10 (b)表明多孔镍是由大量的镍小球(300~500 nm)组成的镍大球(2 μm)构成,该结构一方面显著增加比表面积,另一方面镍小球与镍小球之间存在大量的间隙,能够增强传质能力.同时,从图10 (b)还可观测到NiFe LDH纳米片均匀生长在多孔镍的表面.EIS测试结果表明该多级结构能够显著加快传质过程并能够减小电荷在电极-电解液界面的转移阻力.此外,接触测试结果表明NiFe/Ni/Ni催化电极的液体接触角为0°,表现出超亲水性质,为此电解液能够快速渗入到活性位点,确保活性位点被电解液充分浸润.同时其水下气体接触角为158.7°,具有超疏气性质,为此在电极表面产生的气体气泡能够快速脱附,确保有效的工作面积和稳定的三相活性位点.为此,NiFe/Ni/Ni催化电极具有高传质能力、高电荷转移速度和高结构稳定性等优点,从而表现出优异的催化性能.在10和500 mA/cm2 的电流密度下所需的析氧过电位分别为190和300 mV[图10 (c)],同时也表现出高催化析氢性能[图10 (d)].使用NiFe/Ni/Ni催化电极作为阳极和阴极用于全电解水,在1.96 V的电解电压下就能到达500 mA/cm2 ,并能在500 mA/cm2 下长时间稳定工作. ...
... [
53 ]
(a) The low-magnification SEM image of NiFe/Ni/Ni; (b) The high-magnification SEM imageof NiFe/Ni/Ni; (c) OER performance; (d) HER performance<sup>[<xref ref-type="bibr" rid="R53">53</xref>]</sup> Fig. 10 ![]()
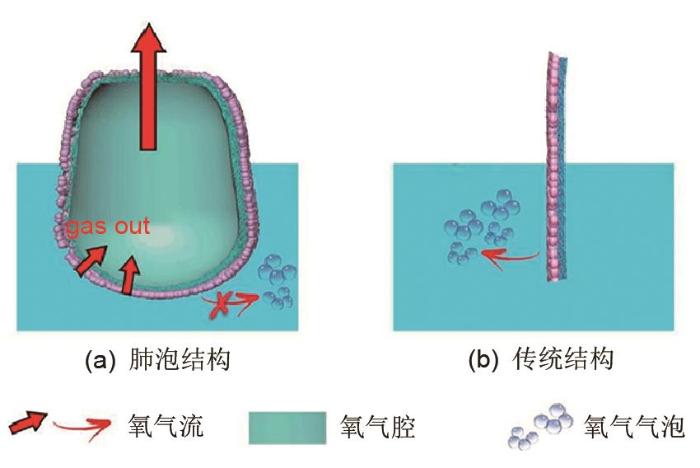
除此之外,还可以通过设计电极结构来实现传质强化.Li等[54 ] 报道了一种肺泡结构的催化电极,如图11 (a)所示,该结构能够实现气相产物与液相反应物分离.该肺泡结构电极的内侧为聚乙烯薄膜,外侧为金薄膜支撑的NiFeO x x 图11 (b)]的催化活性,其在电流密度为10 mA/cm2 时,所需的析氧过电位仅仅为190 mV,性能优于现有研究报道. ...
... [
53 ]
Fig. 10 ![]()
除此之外,还可以通过设计电极结构来实现传质强化.Li等[54 ] 报道了一种肺泡结构的催化电极,如图11 (a)所示,该结构能够实现气相产物与液相反应物分离.该肺泡结构电极的内侧为聚乙烯薄膜,外侧为金薄膜支撑的NiFeO x x 图11 (b)]的催化活性,其在电流密度为10 mA/cm2 时,所需的析氧过电位仅仅为190 mV,性能优于现有研究报道. ...
3
... 除此之外,还可以通过设计电极结构来实现传质强化.Li等[54 ] 报道了一种肺泡结构的催化电极,如图11 (a)所示,该结构能够实现气相产物与液相反应物分离.该肺泡结构电极的内侧为聚乙烯薄膜,外侧为金薄膜支撑的NiFeO x x 图11 (b)]的催化活性,其在电流密度为10 mA/cm2 时,所需的析氧过电位仅仅为190 mV,性能优于现有研究报道. ...
... [
54 ]
Schematic representation of the OER gas delivery path for (a) an alveolus-like polyethylene structure and (b) a flat structure<sup>[<xref ref-type="bibr" rid="R54">54</xref>]</sup> Fig. 11 ![]()
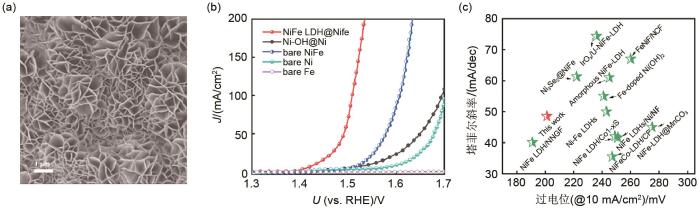
<strong>2.4</strong> 大面积自支撑催化电极的制备 基于对3个关键的认识,课题组发展了自源生长方法(将基底同时作为反应物与导电材料),采用“酸刻蚀/气相生长”法制备自支撑催化析氧电极的绿色工艺方法.将基底元素直接作为反应物可以避免使用外加物质,此时原位生长的催化剂与基底将会有更高的兼容性,催化剂与基底能够实现“一体化”结合.通过酸刻蚀法对泡沫镍铁表面进行原位刻蚀(常温、常压),使用空气中的氧气进行氧化反应,在泡沫镍铁表面原位生长NiFe LDH纳米片阵列[图12 (a)],即自支撑镍铁@镍铁阳极(NiFe LDH@NiFe)[55 ] .NiFe LDH@NiFe催化电极表现出了优异的催化活性,在电流密度为10 mA/cm2 时所需的析氧过电位仅仅为201 mV,远优于大多数文献[55 ] 报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...
... [
54 ]
Fig. 11 ![]()
<strong>2.4</strong> 大面积自支撑催化电极的制备 基于对3个关键的认识,课题组发展了自源生长方法(将基底同时作为反应物与导电材料),采用“酸刻蚀/气相生长”法制备自支撑催化析氧电极的绿色工艺方法.将基底元素直接作为反应物可以避免使用外加物质,此时原位生长的催化剂与基底将会有更高的兼容性,催化剂与基底能够实现“一体化”结合.通过酸刻蚀法对泡沫镍铁表面进行原位刻蚀(常温、常压),使用空气中的氧气进行氧化反应,在泡沫镍铁表面原位生长NiFe LDH纳米片阵列[图12 (a)],即自支撑镍铁@镍铁阳极(NiFe LDH@NiFe)[55 ] .NiFe LDH@NiFe催化电极表现出了优异的催化活性,在电流密度为10 mA/cm2 时所需的析氧过电位仅仅为201 mV,远优于大多数文献[55 ] 报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...
4
... 基于对3个关键的认识,课题组发展了自源生长方法(将基底同时作为反应物与导电材料),采用“酸刻蚀/气相生长”法制备自支撑催化析氧电极的绿色工艺方法.将基底元素直接作为反应物可以避免使用外加物质,此时原位生长的催化剂与基底将会有更高的兼容性,催化剂与基底能够实现“一体化”结合.通过酸刻蚀法对泡沫镍铁表面进行原位刻蚀(常温、常压),使用空气中的氧气进行氧化反应,在泡沫镍铁表面原位生长NiFe LDH纳米片阵列[图12 (a)],即自支撑镍铁@镍铁阳极(NiFe LDH@NiFe)[55 ] .NiFe LDH@NiFe催化电极表现出了优异的催化活性,在电流密度为10 mA/cm2 时所需的析氧过电位仅仅为201 mV,远优于大多数文献[55 ] 报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...
... [55 ]报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...
... [
55 ]
: (a) SEM 图;
(b) 析氧活性;
(c) 性能对比图
NiFe LDH@NiFe<sup>[<xref ref-type="bibr" rid="R55">55</xref>]</sup> (a) SEM image; (b) OER performance; (c) the comparison of catalytic activity Fig. 12 ![]()
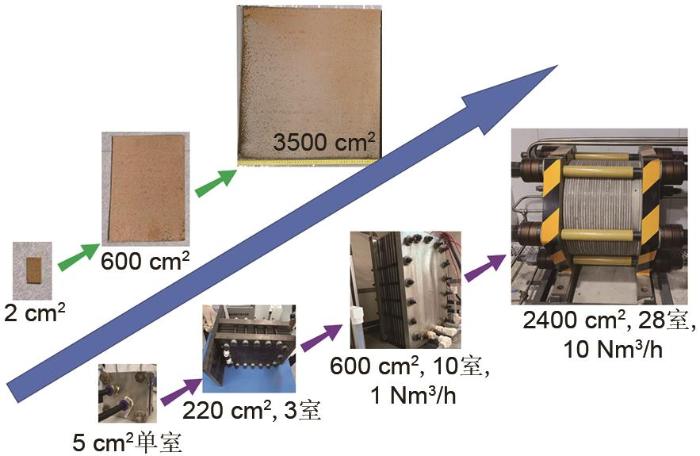
基于“酸刻蚀/气相生长”法制备纳米界面的工程原理,在给定温度、湿度条件下,使用空气中的氧气进行氧化反应,就能够在泡沫镍铁表面原位生长纳米片阵列,课题组完成自支撑催化电极工程方法过程,所制备的面积达到3500 cm2 .在电极表面不同位置取样测定,验证催化性能与电极结构的均匀性.与自主开发的自支撑结构的催化析氢电极配合,使用6 mol/L氢氧化钾水溶液中进行电解水制氢,经过1200 h连续运行,验证自制催化电极和制氢装置长期操作稳定性.将其用于每小时产氢10 Nm3 /h的碱性水溶液电解槽测试(图13 ),电流密度3729 A/m2 条件下,制氢能耗为4.5 kWh/Nm3 H2 ,达到1级国标规定的能效水平. ...
... [
55 ] (a) SEM image; (b) OER performance; (c) the comparison of catalytic activity
Fig. 12 ![]()
基于“酸刻蚀/气相生长”法制备纳米界面的工程原理,在给定温度、湿度条件下,使用空气中的氧气进行氧化反应,就能够在泡沫镍铁表面原位生长纳米片阵列,课题组完成自支撑催化电极工程方法过程,所制备的面积达到3500 cm2 .在电极表面不同位置取样测定,验证催化性能与电极结构的均匀性.与自主开发的自支撑结构的催化析氢电极配合,使用6 mol/L氢氧化钾水溶液中进行电解水制氢,经过1200 h连续运行,验证自制催化电极和制氢装置长期操作稳定性.将其用于每小时产氢10 Nm3 /h的碱性水溶液电解槽测试(图13 ),电流密度3729 A/m2 条件下,制氢能耗为4.5 kWh/Nm3 H2 ,达到1级国标规定的能效水平. ...
1
... 基于对3个关键的认识,课题组发展了自源生长方法(将基底同时作为反应物与导电材料),采用“酸刻蚀/气相生长”法制备自支撑催化析氧电极的绿色工艺方法.将基底元素直接作为反应物可以避免使用外加物质,此时原位生长的催化剂与基底将会有更高的兼容性,催化剂与基底能够实现“一体化”结合.通过酸刻蚀法对泡沫镍铁表面进行原位刻蚀(常温、常压),使用空气中的氧气进行氧化反应,在泡沫镍铁表面原位生长NiFe LDH纳米片阵列[图12 (a)],即自支撑镍铁@镍铁阳极(NiFe LDH@NiFe)[55 ] .NiFe LDH@NiFe催化电极表现出了优异的催化活性,在电流密度为10 mA/cm2 时所需的析氧过电位仅仅为201 mV,远优于大多数文献[55 ] 报道[图12 (b)、(c)].研究表明NiFe LDH纳米片阵列垂直生长在泡沫镍铁表面,HR-TEM测试结果进一步说明NiFe LDH与NiFe合金实现原子级别上的紧密结合,证明了催化剂与基底间具有较完美的固/固界面,这个特征将有助于增强催化剂与基底的结合力,能显著增强催化电极的稳定性[32 , 56 ] .同时,电化学结果分析证明NiFe LDH@NiFe催化电极活性增强主要归因于NiFe LDH催化剂与NiFe合金金属之间的高度匹配性. 此外,接触角测试表明NiFe LDH@NiFe具有超亲水性和优异的疏气性质,有助于电解液充分浸润活性位点并使得产生的氧气气泡快速脱附,具有优异的催化电极-电解液界面. ...




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}