Rapid exploitation of renewable energy sources to replace conventional fossil fuels drives the development of electrical energy storage systems. With the increasing demand for grid-scale energy storage systems, potassium-ion batteries (PIBs) have emerged as a promising alternative to commercial lithium-ion batteries owing to their low cost, natural abundance of potassium resources, low standard reduction potential of potassium, and fascinating transport kinetics of the K+ ions in the electrolyte. Owing to the high abundance of potassium and their low cost, PIBs have considerable advantages in secondary battery energy storage systems. The main challenge in the commercialization of PIBs is finding suitable cathode materials with fast transport kinetics and stable framework structures to intercalate/de-intercalate large-size K+ ions. Transition metal layered oxides have excellent potential and have been extensively investigated as cathode materials for PIBs because of their stable skeleton structure, simple synthetic chemistry, and low cost. In this paper, the effects of the potassium content and synthesis temperature on the crystal structure of transition metal layered oxides are introduced and the structural evolution and capacity loss mechanisms of various crystal structures during potassium removal are explained. Furthermore, modification methods for Mn-based transition metal layered oxides with different crystal structures are proposed to improve their electrochemical properties. Finally, the main research directions for novel transition metal layered oxide cathodes are discussed to provide guidelines for the development of advanced PIBs.
Keywords:failure mechanism
;
element doping
;
surface coating
;
PIBs
;
layered transition metal oxides
HAN Wenzhe. Advances toward manganese-based layered oxide cathodes for potassium-ion batteries[J]. Energy Storage Science and Technology, 2023, 12(5): 1364-1379
Fig. 1
(a) Schematic illustration of the cell configuration and operational mechanism of typical full PIBs[11]; (b) Comparison of the physical and electrochemical properties of Li, Na and K; (c) Electrochemical performance comparison of inorganic cathode materials for PIBs[11]
尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战。二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的。但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后。例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变。因此,在充电/放电曲线中通常存在一系列特征平台。随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视。因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的。寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急。在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料。这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力。近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多。报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17]。图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较。普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能。有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用。聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配。而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20]。2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21]。锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出。但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点。与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理。在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等。最后,对未来钾离子电池领域的主要研究方向和热点进行了展望。总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解。
1 钾含量和合成温度等对锰基层状氧化物晶体结构的影响
Liu等[22]通过简单的共沉淀法制备了钾离子电池锰基层状K x MnO2(x=0.3和0.45)P2/P3型正极材料。结果表明,K的加入量不仅对样品的结构和形貌有重要影响,而且对电化学性能也有一定的影响。在相同的合成条件下,K0.45MnO2样品的结构与K0.3MnO2样品不同,锰基过渡金属层状氧化物颗粒的大小受K含量的影响,K含量较高(x=0.45)的颗粒尺寸较小。相反,当K含量降低时,颗粒尺寸增大。值得注意的是,P3-K0.5MnO2的电压剖面与K0.3MnO2的电压剖面具有相同的特征,其结构为P2型层状,呈现正交畸变。这表明,电势是由K+层中的K含量和K+/空位顺序等分布决定的,而不是由MnO2堆积的多态性决定的。在绝大多数情况下,过渡金属元素与周围六个氧形成的MO6八面体结构组成过渡金属层,钾离子位于过渡金属层之间,形成MO6多面体层与KO6碱金属层交替排布的层状结构。根据MO6多面体中钾离子的配位构型与氧的堆垛方式,将层状氧化物分为O3、P3和P2等不同结构,其中大写的英文字母代表钾离子的配位构型(O是Octahedral的缩写,即八面体位置:P为Prismatic的缩写,即三棱柱位置),数字代表氧最少重复单元的堆垛层数(2对应ABBA……,3对应ABCABC……),人们就对P2、P3和O3等不同相的制备方法进行了深入的研究,发现与P2相(650~900 ℃)相比,P3和O3相可以在较低的煅烧温度(500~550 ℃)下合成,P3相也可以通过TMO2滑动而不破坏其TM—O键从O3相转变来制得[23]。
Fig. 2
(a) Schematic illustrations of the crystal structures of O3-A1-x MeO2, P3-A1-x MeO2, and P2-A1-x MeO2(A=K)[11]; (b) Band structures between the M and ligand orbitals[24]; (c) O 2p nonbonding states and three cases of cationic and anionic redox[24]
P2型K x TMO2结构具有沿c轴的ABBA氧堆叠,而其TMO2层可以标为具有P63/mmc空间群的六边形对称结构。在P2结构中,KO6三棱柱分为两种,一种是三棱柱上下两侧均与过渡金属MO6八面体以共棱形式连接,另外一种是上下两侧均与过渡金属MO6八面体以共面形式连接,这两种钾位分别称为Ke(edge,共棱连接,2d位)和Kf(face,共面连接,2b位)。对于P2结构如果Ke和Kf位置同时占满,钾含量可以达到2,但是由于存在较强的库仑斥力,两个相邻的位置不能同时被占据,一段Ke位置相对Kf位置能量更低,更易被占据,但是两个位置的K+占据比例与充放电状态和过渡金属元素的选择均有关系。
Fig. 3
(a) Enlarged HRTEM image of K0.77MnO2 ⋅H2O0.23[26]; (b) The first galvanostatic charge/discharge curves of the K0.77MnO2 ⋅H2O0.23 electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2θ region of 10.5°~15.5°[26]; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles[26]; (d) In situ XRD characterization of K0.27(Mn0.98O2)⋅(H2O)0.53 electrode upon charge/discharge[43]; (e) Operando-XRD result of P2-K0.75[Ni1/3Mn2/3]O2 electrode[44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]
一般情况下,P3-K x MnO2正极材料在充放电过程中会发生多相转变,在脱钾状态下向O3型层状结构转变,从而引起较大的体积变化。这是由于钾位空位形成的氧-氧排斥作用的结果。这可能是容量衰减的原因,也同样导致了具有这种现象的样品在电化学测试时呈现出非平滑的充放电曲线。例如,Kim等[48]报道了P3-K0.5MnO2,这项工作可以帮助大家更好地理解层状P3型K0.5MnO2中的晶体结构演变,并且对钾离子电池新型正极材料的设计和开发提供了见解。图4(a)展示了P3-K0.5MnO2正极在2 mA/g电流密度下循环时收集的原位XRD图谱。我们观察到在特定K含量(K x MnO2中的0.395<x<0.425和0.316<x<0.364)与其他K组成的固溶态的两相反应。K脱嵌后,(003)和(006)峰位移到较低的角度[图4(b)、(c)],表明MnO2平板距离扩大。更重要的是,在K x MnO2中,(015)峰消失,在x≈0.41处出现一个新的(104)峰。这一特征表明随着钾离子的去除,从P3结构转变为O3结构[49]。并且,当K x MnO2进一步脱嵌K至x≈0.34后,在(003)和(006)峰的左边出现了一组新的XRD峰,如图4(b)、(c)所示,表明发生了另一个相变。在1.5~4.0 V的工作电压窗口中,在电流密度为16 mA/g时,其初始比容量为140 mAh/g。即使在如此低的截止电压下测试,通过原位X射线(XRD)表征也观察到的P3-O3相变,这应该是循环性能下降的原因。
图4
(a) P3型K0.5MnO2 在电流密度为2 mA/g时的充放电曲线;(b)~(d) #1、#7和#14的原位XRD图;(e) P3-K0.5MnO2 的XRD峰比较;(f) O3和P3结构的XRD模拟图谱比较[48];(g) P3-K0.75MnO2 和P3-K0.75[Co0.5Mn0.5]O2 的晶体结构示意图;(h) P3-K x [Co0.5Mn0.5]O2(0.25≤ x ≤0.75)的结构变化;(i) P3-K x [Co0.5Mn0.5]O2 的XRD谱图;(j) 通过DFT计算和XRD数据得到的P3-K x [Co0.5Mn0.5]O2 的晶格参数的对比图[57];(k) 花生状P3型K0.45Mn0.5Co0.5O2 微粒的合成示意图[59];(l)、(m) P3型K0.45Mn0.5Co0.5O2 的SEM图像[59];(n) P3型K0.45Mn0.5Co0.5O2 在300 mA/g电流密度下的长期循环性能[59];(o) K0.5MnO2 的循环伏安图
Fig. 4
(a) Typical charge/discharge profiles of P3-type K0.5MnO2 at a current rate of 2 mA/g; (b)~(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K0.5MnO2 at 18°~20°; (f) comparison with simulated XRD patterns of the O3 and P3structures[48]; (g) P3-K0.75MnO2 and P3-K0.75[Co0.5Mn0.5]O2; (h) Predicted structural change in P3-K x [Co0.5Mn0.5]O2 (0.25≤ x ≤0.75); (i) operando XRD patterns of P3-K x [Co0.5Mn0.5]O2; (j) comparison of c-lattice parameters of P3-K x [Co0.5Mn0.5]O2 obtained by first-principles calculations and operando XRD data[57]; (k) Schematic illustration of the synthesis of peanut-like P3-type K0.45Mn0.5Co0.5O2 microparticles[59]; (l), (m) SEM images of P3-type K0.45Mn0.5Co0.5O2; (n) long-term cycling performance of K0.45Mn0.5Co0.5O2 at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸。据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50]。这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因。这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51]。因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱。
3.2.3 结构改性
与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性。如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响。由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化。利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变。除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)]。这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变。这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布。预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)]。基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持。实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K)。在20 mA/g下超过100次循环,可保持其初始容量的85%。即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能。
研究人员还发现传统的样品制备方法,例如固相方法,通常会产生不规则形状的颗粒,其表面结构容易被破坏,并逐渐渗透到本体相中,导致库仑效率低,容量衰减快。而相较下结构工程由于其优异的结构稳定性,可以对抗连续嵌入/脱嵌钾离子引起的机械应力而得到了很好的设计研究。控制合成具有良好形貌的层状氧化物材料,既能提高结构稳定性,又能抑制副反应,有利于充分发挥层状锰基正极结构的K+存储潜力[58]。最近,报道了P3-K0.45Mn0.5Co0.5O2[59]。Co占据Mn位点,以抑制Mn3+的Jahn-Teller效应,并且独特的花生状结构[图4(k)~(m)]不仅大大缩短了K+的扩散距离,还缓解了K+连续嵌入/脱嵌引起的内部应变,从而改善了反应动力学和结构稳定性,提高了P3-K0.45Mn0.5Co0.5O2的循环稳定性和倍率性能。这为实现令人满意的钾离子电池晶格结构设计提供了新的策略。其在电流密度为300 mA/g的条件下,具有稳定的循环能力,在300次循环后保持了73.8%的高比容量[图4(n)],之后仍然保持中空特征和高结晶度,没有任何杂质,证实了其突出的结构稳定性。值得注意的是,上述P3型锰基层状氧化物的电压窗口都未超过4 V,这是因为降低截止电压对于稳定P3-K x MnO2的层状结构也有帮助。例如[48]在1.5~4.2 V (vs K/K+)扫描时,在约3.7 V和4.1 V(vs K/K+)处观察到两个强氧化峰,而对应的还原峰不太明显[图4(o)]。相反,在1.5~3.9 V扫描时,氧化峰和还原峰匹配良好,这些结果表明,K的脱嵌和嵌入过程可能导致高电压区发生不可逆的结构变化。因此,降低截止电压对于提高P3-K x MnO2的层状结构在充放电过程中的可逆性具有积极影响。
Fig. 5
(a) In situ XRD characterization of K0.5Mn0.6Co0.2Fe0.1Mg0.1O2[61]; (b) Visualization of Jahn-Teller distortion[61]; (c) Long-term cycling performance of K0.5Mn0.6Co0.2Fe0.1Mg0.1O2 at 1 A/g[61]; (e) In situ XRD patterns of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (f) HRTEM images of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略。因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题。在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)]。设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能。在20 mA/g时具有117 mAh/g的可逆钾存储容量。原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性。在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化。随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动。峰位的逐渐变化归因于结构沿c轴方向的膨胀。Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性。P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化。这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩。在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因。
Fig. 6
(a) Schematic diagram of the effect of Rb and Mg substitution on the crystal structure; (b) The stacking of P3-type and O3-type layered structures along the c-axis direction of the TM slabs; (c) Representative HRTEM images of the K0.45Rb0.05Mn0.85Mg0.15O2 powder and K0.45Rb0.05Mn0.85Mg0.15O2 test cathode placed in the battery environment without cycling; (d) The voltage curves and representative corresponding in situ XRD patterns of K0.45MnO2 and K0.45Rb0.05Mn0.85Mg0.15O2 at 70 mA/g in the range of 1.5—3.9 V.; (e) The c-lattice parameter of K0.45MnO2 and K0.45Rb0.05Mn0.85Mg0.15O2 calculated from in situ XRD patterns; (f) Charge GITT result of K0.45Rb0.05Mn0.85Mg0.15O2 at 20 mA/g, and the corresponding DK+; (g) Cycling performance of K0.45Rb0.05Mn0.85Mg0.15O2 at 200 mA/g[69]
PRAMUDITA J C, SEHRAWAT D, GOONETILLEKE D, et al. An initial review of the status of electrode materials for potassium-ion batteries[J]. Advanced Energy Materials, 2017, 7(24): 1602911.
XU Y S, DUAN S Y, SUN Y G, et al. Recent developments in electrode materials for potassium-ion batteries[J]. Journal of Materials Chemistry A, 2019, 7(9): 4334-4352.
WANG J, LIU Z M, ZHOU J, et al. Insights into metal/metalloid-based alloying anodes for potassium ion batteries[J]. ACS Materials Letters, 2021, 3(11): 1572-1598.
HAN K, MENG J S, HONG X F, et al. Three-dimensional graphene-supported nickel disulfide nanoparticles promise stable and fast potassium storage[J]. Nanoscale, 2020, 12(15): 8255-8261.
WANG X P, XIAO Z T, HAN K, et al. Advances in fine structure optimizations of layered transition-metal oxide cathodes for potassium-ion batteries[J]. Advanced Energy Materials, 2023, 13(2): 2202861.
LI J Y, MANTHIRAM A. A comprehensive analysis of the interphasial and structural evolution over long-term cycling of ultrahigh-nickel cathodes in lithium-ion batteries[J]. Advanced Energy Materials, 2019, 9(45): 1902731.
HWANG J Y, KIM J, YU T Y, et al. A new P2-type layered oxide cathode with superior full-cell performances for K-ion batteries[J]. Journal of Materials Chemistry A, 2019, 7(37): 21362-21370.
XU Y S, ZHANG Q H, WANG D, et al. Enabling reversible phase transition on K5/9Mn7/9Ti2/9O2 for high-performance potassium-ion batteries cathodes[J]. Energy Storage Materials, 2020, 31: 20-26.
ZHANG L, ZHANG B W, WANG C R, et al. Constructing the best symmetric full K-ion battery with the NASICON-type K3V2(PO4)3[J]. Nano Energy, 2019, 60: 432-439.
CHONG S K, WU Y F, GUO S W, et al. Potassium nickel hexacyanoferrate as cathode for high voltage and ultralong life potassium-ion batteries[J]. Energy Storage Materials, 2019, 22: 120-127.
KAPAEV R R, ZHIDKOV I S, KURMAEV E Z, et al. Hexaazatriphenylene-based polymer cathode for fast and stable lithium-, sodium- and potassium-ion batteries[J]. Journal of Materials Chemistry A, 2019, 7(39): 22596-22603.
KIM U H, JUN D W, PARK K J, et al. Pushing the limit of layered transition metal oxide cathodes for high-energy density rechargeable Li ion batteries[J]. Energy & Environmental Science, 2018, 11(5): 1271-1279.
ZHANG H Y, XI K Y, JIANG K Z, et al. Enhanced K-ion kinetics in a layered cathode for potassium ion batteries[J]. Chemical Communications, 2019, 55(55): 7910-7913.
ZHANG H, WANG L, HE X M. Trends in a study on thermal runaway mechanism of lithium-ion battery with LiNixMnyCo1-x-yO2 cathode materials[J]. Battery Energy, 2022, 1(1): 20210011.
VAALMA C, GIFFIN G A, BUCHHOLZ D, et al. Non-aqueous K-ion battery based on layered K0.3MnO2 and hard carbon/carbon black[J]. Journal of the Electrochemical Society, 2016, 163(7): A1295-A1299.
LIU C L, LUO S H, HUANG H B, et al. Layered potassium-deficient P2- and P3-type cathode materials KxMnO2 for K-ion batteries[J]. Chemical Engineering Journal, 2019, 356: 53-59.
PATOUX S, DOLLÉ M, DOEFF M M. Layered Manganese oxide intergrowth electrodes for rechargeable lithium batteries. 2. substitution with Al[J]. Chemistry of Materials, 2005, 17(5): 1044-1054.
HUANG Z X, GU Z Y, HENG Y L, et al. Advanced layered oxide cathodes for sodium/potassium-ion batteries: Development, challenges and prospects[J]. Chemical Engineering Journal, 2023, 452: 139438.
SUN R, DONG S Y, XU F, et al. Co-intercalation strategy of constructing partial cation substitution of ammonium vanadate {(NH4)2V6O16} for stable zinc ion storage[J]. Dalton Transactions, 2022, 51(19): 7607-7612.
LIN B W, ZHU X H, FANG L Z, et al. Birnessite nanosheet arrays with high K content as a high-capacity and ultrastable cathode for K-ion batteries[J]. Advanced Materials, 2019, 31(24): 1900060.
KUMAKURA S, TAHARA Y, SATO S, et al. P'2-Na2/3Mn0.9Me0.1O2 (Me=Mg, Ti, Co, Ni, Cu, and Zn): Correlation between orthorhombic distortion and electrochemical property[J]. Chemistry of Materials, 2017, 29(21): 8958-8962.
BILLAUD J, SINGH G, ARMSTRONG A R, et al. Na0.67Mn1-xMgxO2 (0≤x≤0.2): A high capacity cathode for sodium-ion batteries[J]. Energy & Environmental Science, 2014, 7(4): 1387-1391.
NAM K W, KIM S, YANG E, et al. Critical role of crystal water for a layered cathode material in sodium ion batteries[J]. Chemistry of Materials, 2015, 27(10): 3721-3725.
JO J H, CHOI J U, KONAROV A, et al. Sodium-ion batteries: Building effective layered cathode materials with long-term cycling by modifying the surface via sodium phosphate[J]. Advanced Functional Materials, 2018, 28(14): 1705968.
LIU Z M, WANG J, JIA X X, et al. Graphene armored with a crystal carbon shell for ultrahigh-performance potassium ion batteries and aluminum batteries[J]. ACS Nano, 2019, 13(9): 10631-10642.
BAO S, LUO S H, LU J L. Preparation and optimization of ZrO2 modified P2-type Na2/3Ni1/6Co1/6Mn2/3O2 with enhanced electrochemical performance as cathode for sodium ion batteries[J]. Ceramics International, 2020, 46(10): 16080-16087.
YU Y, KONG W J, LI Q Y, et al. Understanding the multiple effects of TiO2 coating on NaMn0.33Fe0.33Ni0.33O2 cathode material for Na-ion batteries[J]. ACS Applied Energy Materials, 2020, 3(1): 933-942.
ZHANG Y, LIU L, JAMIL S, et al. Al2O3 coated Na0.44MnO2 as high-voltage cathode for sodium ion batteries[J]. Applied Surface Science, 2019, 494: 1156-1165.
SUN H H, HWANG J Y, YOON C S, et al. Capacity degradation mechanism and cycling stability enhancement of AlF3-coated nanorod gradient Na[Ni0.65Co0.08Mn0.27]O2 cathode for sodium-ion batteries[J]. ACS Nano, 2018, 12(12): 12912-12922.
QI X G, WANG W G, HU Y S, et al. Surface modification research of layered oxide materials for sodium-ion batteries[J]. Energy Storage Science and Technology, 2020, 9(5)1396-1401
ZHENG J M, GU M, XIAO J, et al. Functioning mechanism of AlF3 coating on the Li- and Mn-rich cathode materials[J]. Chemistry of Materials, 2014, 26(22): 6320-6327.
ZHAO S Q, YAN K, MUNROE P, et al. Construction of hierarchical K1.39Mn3O6 spheres via AlF3 coating for high-performance potassium-ion batteries[J]. Advanced Energy Materials, 2019, 9(10): 1803757.
TAPIA-RUIZ N, DOSE W M, SHARMA N, et al. High voltage structural evolution and enhanced Na-ion diffusion in P2-Na2/3Ni1/3-xMgxMn2/3O2 (0≤x≤0.2) cathodes from diffraction, electrochemical and ab initio studies[J]. Energy & Environmental Science, 2018, 11(6): 1470-1479.
PIAO J Y, GU L, WEI Z X, et al. Phase control on surface for the stabilization of high energy cathode materials of lithium ion batteries[J]. Journal of the American Chemical Society, 2019, 141(12): 4900-4907.
WANG Q C, MENG J K, YUE X Y, et al. Tuning P2-structured cathode material by Na-site Mg substitution for Na-ion batteries[J]. Journal of the American Chemical Society, 2019, 141(2): 840-848.
GAO A, LI M, GUO N N, et al. K-birnessite electrode obtained by ion exchange for potassium-ion batteries: Insight into the concerted ionic diffusion and K storage mechanism[J]. Advanced Energy Materials, 2019, 9(1): 1802739.
JO J H, CHOI J U, PARK Y J, et al. P2-K0.75[Ni1/3Mn2/3]O2 cathode material for high power and long life potassium-ion batteries[J]. Advanced Energy Materials, 2020, 10(7): 1903605.
WANG S H, SUN C L, WANG N, et al. Ni- and/or Mn-based layered transition metal oxides as cathode materials for sodium ion batteries: Status, challenges and countermeasures[J]. Journal of Materials Chemistry A, 2019, 7(17): 10138-10158.
MA C Z, ALVARADO J, XU J, et al. Exploring oxygen activity in the high energy P2-type Na0.78Ni0.23Mn0.69O2 cathode material for Na-ion batteries[J]. Journal of the American Chemical Society, 2017, 139(13): 4835-4845.
LIU H Q, GAO X, CHEN J, et al. Layered oxide cathode for sodium ion batteries: Interlayer glide, phase transition and performance[J]. Energy Storage Science and Technology, 2020, 9(5): 1327-1339
KIM H, SEO D H, KIM J C, et al. Investigation of potassium storage in layered P3-type K0.5MnO2 cathode[J]. Advanced Materials, 2017, 29(37): 1702480.
BEZZA I, KAUS M, HEINZMANN R, et al. Mechanism of the delithiation/lithiation process in LiFe0.4Mn0.6PO4: in situ and ex situ investigations on long-range and local structures[J]. The Journal of Physical Chemistry C, 2015, 119(17): 9016-9024.
CHOI J U, YOON C S, ZHANG Q, et al. Understanding on the structural and electrochemical performance of orthorhombic sodium Manganese oxides[J]. Journal of Materials Chemistry A, 2019, 7(1): 202-211.
WANG X P, XU X M, NIU C J, et al. Earth abundant Fe/Mn-based layered oxide interconnected nanowires for advanced K-ion full batteries[J]. Nano Letters, 2017, 17(1): 544-550.
CHO M K, JO J H, CHOI J U, et al. Cycling stability of layered potassium Manganese oxide in nonaqueous potassium cells[J]. ACS Applied Materials & Interfaces, 2019, 11(31): 27770-27779.
SADA K, BARPANDA P. P3-type layered K0.48Mn0.4Co0.6O2: A novel cathode material for potassium-ion batteries[J]. Chemical Communications, 2020, 56(15): 2272-2275.
WENG J Y, DUAN J, SUN C L, et al. Construction of hierarchical K0.7Mn0.7Mg0.3O2 microparticles as high capacity & long cycle life cathode materials for low-cost potassium-ion batteries[J]. Chemical Engineering Journal, 2020, 392: 123649.
RAMASAMY H V, SENTHILKUMAR B, BARPANDA P, et al. Superior potassium-ion hybrid capacitor based on novel P3-type layered K0.45Mn0.5Co0.5O2 as high capacity cathode[J]. Chemical Engineering Journal, 2019, 368: 235-243.
CHOI J U, KIM J, HWANG J Y, et al. K0.54[Co0.5Mn0.5]O2: New cathode with high power capability for potassium-ion batteries[J]. Nano Energy, 2019, 61: 284-294.
LIU Z M, WANG J, LU B G. Plum pudding model inspired KVPO4F@3DC as high-voltage and hyperstable cathode for potassium ion batteries[J]. Science Bulletin, 2020, 65(15): 1242-1251.
ZHANG Z Z, SUN J L, DUAN L P, et al. Self-templated construction of peanut-like P3-type K0.45Mn0.5Co0.5O2 for highly reversible potassium storage[J]. Journal of Materials Chemistry A, 2022, 10(2): 554-560.
BAI P L, JIANG K Z, ZHANG X P, et al. Ni-doped layered Manganese oxide as a stable cathode for potassium-ion batteries[J]. ACS Applied Materials & Interfaces, 2020, 12(9): 10490-10495.
XIAO Z T, XIA F J, XU L H, et al. Suppressing the jahn-teller effect in Mn-based layered oxide cathode toward long-life potassium-ion batteries[J]. Advanced Functional Materials, 2022, 32(14): 2108244.
LIU W, LI X F, XIONG D B, et al. Significantly improving cycling performance of cathodes in lithium ion batteries: The effect of Al2O3 and LiAlO2 coatings on LiNi0.6Co0.2Mn0.2O2[J]. Nano Energy, 2018, 44: 111-120.
CHOI J U, KIM J, JO J H, et al. Facile migration of potassium ions in a ternary P3-type K0.5[Mn0.8Fe0.1Ni0.1]O2 cathode in rechargeable potassium batteries[J]. Energy Storage Materials, 2020, 25: 714-723.
XU Y S, ZHOU Y N, ZHANG Q H, et al. Layered oxides with solid-solution reaction for high voltage potassium-ion batteries cathode[J]. Chemical Engineering Journal, 2021, 412: 128735.
ZHANG X Y, YU D X, WEI Z X, et al. Layered P3-type K0.4Fe0.1Mn0.8Ti0.1O2 as a low-cost and zero-strain electrode material for both potassium and sodium storage[J]. ACS Applied Materials & Interfaces, 2021, 13(16): 18897-18904.
LV J R, WANG B, HAO J X, et al. Single-crystalline Mn-based oxide as a high-rate and long-life cathode material for potassium-ion battery[J]. eScience, 2023, 3(1): 100081.
WANG P F, XIN H S, ZUO T T, et al. An abnormal 3.7 Volt O3-Type sodium-ion battery cathode[J]. Angewandte Chemie International Edition, 2018, 57(27): 8178-8183.
DENG J Q, LUO W B, LU X, et al. High energy density sodium-ion battery with industrially feasible and air-stable O3-Type layered oxide cathode[J]. Advanced Energy Materials, 2018, 8(5): 1701610.
CAIXIANG Z M, HAO J X, ZHOU J, et al. Interlayer-engineering and surface-substituting Manganese-based self-evolution for high-performance potassium cathode[J]. Advanced Energy Materials, 2023, 13(1): 2203126.
KIM H, SEO D H, URBAN A, et al. Stoichiometric layered potassium transition metal oxide for rechargeable potassium batteries[J]. Chemistry of Materials, 2018, 30(18): 6532-6539.
JIANG X Y, LIU X W, ZENG Z Q, et al. A nonflammable Na+-based dual-carbon battery with low-cost, high voltage, and long cycle life[J]. Advanced Energy Materials, 2018, 8(36): 1802176.
HUANG Yonghao, ZANG Guojing, ZHU WeiYa, et al. Improvement in interfacial stability between lithium-containing ceramic separator and 4.35 V LiNi0.8Co0.1Mn0.1O2 cathode by LiF additives[J]. Energy Storage Science and Technology, doi: 10.19799/j.cnki.2095-4239. 2023.0128.
HU Z, HAO J X, SHEN D Y, et al. Electro-spraying/spinning: A novel battery manufacturing technology[J]. Green Energy & Environment, 2022, doi: 10.1016/j.gee.2022.05.004.
(a) Schematic illustration of the cell configuration and operational mechanism of typical full PIBs<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>; (b) Comparison of the physical and electrochemical properties of Li, Na and K; (c) Electrochemical performance comparison of inorganic cathode materials for PIBs<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 1
图1(b)简要比较了锂、钠和钾的性质,钾离子电池的优势主要体现在以下几个方面: ...
... [11](a) Schematic illustration of the cell configuration and operational mechanism of typical full PIBs<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>; (b) Comparison of the physical and electrochemical properties of Li, Na and K; (c) Electrochemical performance comparison of inorganic cathode materials for PIBs<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 1
图1(b)简要比较了锂、钠和钾的性质,钾离子电池的优势主要体现在以下几个方面: ...
... [11]; (b) Comparison of the physical and electrochemical properties of Li, Na and K; (c) Electrochemical performance comparison of inorganic cathode materials for PIBs[11]Fig. 1
<strong>(a) Schematic illustrations of the crystal structures of O3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, P3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, and P2-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>(A=K)</strong><sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup><strong>; (b) Band structures between the M and ligand orbitals</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup><strong>; (c) O 2p nonbonding states and three cases of cationic and anionic redox</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup>Fig. 2
... [11]; (b) Band structures between the M and ligand orbitals[24]; (c) O 2p nonbonding states and three cases of cationic and anionic redox[24]Fig. 2
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
6
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [13]<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
0
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... 尽管钾离子电池具有上述优点,但不可否认,钾离子电池的开发研究在如何设计能够维持理想长循环的电极材料方面面临挑战.二次电池的关键电化学性能参数,如可逆比容量、输出电压、速率能力和循环稳定性主要是由正极材料的固有电化学性质决定的.但由于与Na+相比,K+的半径更大,使得电极材料内部K+的嵌入/脱嵌容易引起晶格结构的大体积变形,从而使材料处于结构失效的高风险中,尤其是在长循环周期之后.例如,含钾的层状氧化物在插入/提取K+时更容易遭受严重的晶格畸变(Δc为4.7%,而锂离子电池的Δc值为3.3%[12-13]),这导致复杂的相变.因此,在充电/放电曲线中通常存在一系列特征平台.随着正极结构塌陷的加重,组装好的电池还会经历持续的容量衰减,这种因钾离子具有较大的离子半径而引起的对电池性能的严重影响不容忽视.因此,直接将锂离子电极材料的结构用在钾离子电池体系中是不合适的.寻找既能提供高水平电化学性能,又能有效抑制电化学过程中结构退化的功能材料成为当务之急.在钾离子电池系统中,主要的研究重点是找到在大尺寸K+的脱嵌/嵌入过程中仍具有坚固框架结构的正极材料.这不仅能为大尺寸K+的快速传输提供大通道,而且即使在高电压范围内也能保持完整的晶格框架,从而实现高容量的长循环能力.近年来,对于钾离子电池正极材料的研究不断增长,开创性文献报道逐渐增多.报道的钾离子电池正极材料分为四类:层状过渡金属氧化物正极材料[14]、聚阴离子类正极材料[15]、普鲁士蓝类正极材料[16]、有机类正极材料[17].图1(c)显示了钾离子无机正极材料关键电化学参数的综合比较.普鲁士蓝类正极材料虽然有较高的比容量,但材料的电子导电性较差,因此很难提高倍率性能.有机电极材料具有可再生、绿色、成本低、容量高等优点,但难以找到含钾却不在电解液中溶解的有机正极材料,极大地限制了此类材料在钾离子电池中的实际应用.聚阴离子化合物通常具有较高的电化学反应稳定性和工作电压,但这种理论上的高循环稳定性难以找到合适的电解液与之匹配.而在这些候选材料中,层状过渡金属氧化物(A x MO2,A=K,Na及其组合;M=V,Mn,Fe,Ni,Cr,Co等及其组合)由于其稳定的框架结构,简单的化学合成方法,高输出电压和商业化生产的可行性而被广泛开发[18-20].2016年,Valma等人开发了层状氧化物K0.3MnO2作为钾离子电池正极材料[21].锰元素作为一种丰富的过渡金属元素,具有容量高、可持续性和成本低的特殊优势,并且Mn基层状过渡金属氧化物因其具有安全性、稳定性和价格优势而在钾离子电池材料的选择中脱颖而出.但和其他所有钾离子电池正极材料一样,Mn基层状过渡金属氧化物在进行结构设计以获得突出的长周期能力时面临着结构稳定性的挑战,因此研究钾离子电池在循环过程中的晶体结构演变和容量损失机理,并以此为基础进一步进行结构改性成为最为重要和本质的研究焦点.与以往对钾离子电池电极材料的综述不同,本文先介绍了钾的加入量和合成温度等对锰基过渡金属层状氧化物结构类型的影响,并分别说明了各类型的电极材料的晶体结构和其在脱钾过程中的晶体结构演变和容量损失机理.在此基础上,分别提出了针对各类锰基过渡金属层状氧化物的改性方法以提高其电化学性能,如表面改性抑制副反应稳定界面、元素取代调整微观结构、调整电压窗口等.最后,对未来钾离子电池领域的主要研究方向和热点进行了展望.总的来说,这篇综述将为钾离子电池锰基层状氧化物正极的研究工作提供见解. ...
1
... Liu等[22]通过简单的共沉淀法制备了钾离子电池锰基层状K x MnO2(x=0.3和0.45)P2/P3型正极材料.结果表明,K的加入量不仅对样品的结构和形貌有重要影响,而且对电化学性能也有一定的影响.在相同的合成条件下,K0.45MnO2样品的结构与K0.3MnO2样品不同,锰基过渡金属层状氧化物颗粒的大小受K含量的影响,K含量较高(x=0.45)的颗粒尺寸较小.相反,当K含量降低时,颗粒尺寸增大.值得注意的是,P3-K0.5MnO2的电压剖面与K0.3MnO2的电压剖面具有相同的特征,其结构为P2型层状,呈现正交畸变.这表明,电势是由K+层中的K含量和K+/空位顺序等分布决定的,而不是由MnO2堆积的多态性决定的.在绝大多数情况下,过渡金属元素与周围六个氧形成的MO6八面体结构组成过渡金属层,钾离子位于过渡金属层之间,形成MO6多面体层与KO6碱金属层交替排布的层状结构.根据MO6多面体中钾离子的配位构型与氧的堆垛方式,将层状氧化物分为O3、P3和P2等不同结构,其中大写的英文字母代表钾离子的配位构型(O是Octahedral的缩写,即八面体位置:P为Prismatic的缩写,即三棱柱位置),数字代表氧最少重复单元的堆垛层数(2对应ABBA……,3对应ABCABC……),人们就对P2、P3和O3等不同相的制备方法进行了深入的研究,发现与P2相(650~900 ℃)相比,P3和O3相可以在较低的煅烧温度(500~550 ℃)下合成,P3相也可以通过TMO2滑动而不破坏其TM—O键从O3相转变来制得[23]. ...
1
... Liu等[22]通过简单的共沉淀法制备了钾离子电池锰基层状K x MnO2(x=0.3和0.45)P2/P3型正极材料.结果表明,K的加入量不仅对样品的结构和形貌有重要影响,而且对电化学性能也有一定的影响.在相同的合成条件下,K0.45MnO2样品的结构与K0.3MnO2样品不同,锰基过渡金属层状氧化物颗粒的大小受K含量的影响,K含量较高(x=0.45)的颗粒尺寸较小.相反,当K含量降低时,颗粒尺寸增大.值得注意的是,P3-K0.5MnO2的电压剖面与K0.3MnO2的电压剖面具有相同的特征,其结构为P2型层状,呈现正交畸变.这表明,电势是由K+层中的K含量和K+/空位顺序等分布决定的,而不是由MnO2堆积的多态性决定的.在绝大多数情况下,过渡金属元素与周围六个氧形成的MO6八面体结构组成过渡金属层,钾离子位于过渡金属层之间,形成MO6多面体层与KO6碱金属层交替排布的层状结构.根据MO6多面体中钾离子的配位构型与氧的堆垛方式,将层状氧化物分为O3、P3和P2等不同结构,其中大写的英文字母代表钾离子的配位构型(O是Octahedral的缩写,即八面体位置:P为Prismatic的缩写,即三棱柱位置),数字代表氧最少重复单元的堆垛层数(2对应ABBA……,3对应ABCABC……),人们就对P2、P3和O3等不同相的制备方法进行了深入的研究,发现与P2相(650~900 ℃)相比,P3和O3相可以在较低的煅烧温度(500~550 ℃)下合成,P3相也可以通过TMO2滑动而不破坏其TM—O键从O3相转变来制得[23]. ...
<strong>(a) Schematic illustrations of the crystal structures of O3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, P3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, and P2-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>(A=K)</strong><sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup><strong>; (b) Band structures between the M and ligand orbitals</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup><strong>; (c) O 2p nonbonding states and three cases of cationic and anionic redox</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup>Fig. 2
... [24]<strong>(a) Schematic illustrations of the crystal structures of O3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, P3-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>, and P2-A<sub>1</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> MeO<sub>2</sub>(A=K)</strong><sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup><strong>; (b) Band structures between the M and ligand orbitals</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup><strong>; (c) O 2p nonbonding states and three cases of cationic and anionic redox</strong><sup>[<xref ref-type="bibr" rid="R24">24</xref>]</sup>Fig. 2
... [26];(b) K0.77MnO2 ⋅H2O0.23 电极在电流密度为20 mA/g时的充放电曲线及其在10.5°~15.5°区域的原位XRD谱图[26];(c) 电流密度为100 mA/g,循环100次时电极的循环性能和相应的库仑效率[26];(d) K0.77MnO2 ⋅H2O0.23 电极充放电时的原位XRD表征[43];(e) P2-K0.75[Ni1/3Mn2/3]O2 电极的XRD结果[44];(f) 相应的计算晶格参数[44];(g) 由实验计算的数据和第一性原理计算预测的数据[13];(h) P2-K0.75MNFO2 在1.5~3.9 V(K/K+)电压范围内充放电过程的XRD图谱[13]<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [26];(c) 电流密度为100 mA/g,循环100次时电极的循环性能和相应的库仑效率[26];(d) K0.77MnO2 ⋅H2O0.23 电极充放电时的原位XRD表征[43];(e) P2-K0.75[Ni1/3Mn2/3]O2 电极的XRD结果[44];(f) 相应的计算晶格参数[44];(g) 由实验计算的数据和第一性原理计算预测的数据[13];(h) P2-K0.75MNFO2 在1.5~3.9 V(K/K+)电压范围内充放电过程的XRD图谱[13]<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [26];(d) K0.77MnO2 ⋅H2O0.23 电极充放电时的原位XRD表征[43];(e) P2-K0.75[Ni1/3Mn2/3]O2 电极的XRD结果[44];(f) 相应的计算晶格参数[44];(g) 由实验计算的数据和第一性原理计算预测的数据[13];(h) P2-K0.75MNFO2 在1.5~3.9 V(K/K+)电压范围内充放电过程的XRD图谱[13]<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [26]; (b) The first galvanostatic charge/discharge curves of the K0.77MnO2 ⋅H2O0.23 electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2θ region of 10.5°~15.5°[26]; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles[26]; (d) In situ XRD characterization of K0.27(Mn0.98O2)⋅(H2O)0.53 electrode upon charge/discharge[43]; (e) Operando-XRD result of P2-K0.75[Ni1/3Mn2/3]O2 electrode[44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
... [26]; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles[26]; (d) In situ XRD characterization of K0.27(Mn0.98O2)⋅(H2O)0.53 electrode upon charge/discharge[43]; (e) Operando-XRD result of P2-K0.75[Ni1/3Mn2/3]O2 electrode[44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
... [26]; (d) In situ XRD characterization of K0.27(Mn0.98O2)⋅(H2O)0.53 electrode upon charge/discharge[43]; (e) Operando-XRD result of P2-K0.75[Ni1/3Mn2/3]O2 electrode[44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [43]; (e) Operando-XRD result of P2-K0.75[Ni1/3Mn2/3]O2 electrode[44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [44];(g) 由实验计算的数据和第一性原理计算预测的数据[13];(h) P2-K0.75MNFO2 在1.5~3.9 V(K/K+)电压范围内充放电过程的XRD图谱[13]<strong>(a) Enlarged HRTEM image of K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub></strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (b) The first galvanostatic charge/discharge curves of the K<sub>0.77</sub>MnO<sub>2</sub></strong> ⋅<strong>H<sub>2</sub>O<sub>0.23</sub> electrode at a current density of 20 mA/g and corresponding ex situ XRD patterns in the 2<i>θ</i> region of 10.5°</strong>~<strong>15.5°</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (c) Cycle performances and corresponding Coulombic efficiencies of the electrode at a current density of 100 mA/g for 100 cycles</strong><sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup><strong>; (d) In situ XRD characterization of K<sub>0.27</sub>(Mn<sub>0.98</sub>O<sub>2</sub>)</strong>⋅<strong>(H<sub>2</sub>O)<sub>0.53</sub> electrode upon charge/discharge</strong><sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup><strong>; (e) Operando-XRD result of P2-K<sub>0.75</sub>[Ni<sub>1/3</sub>Mn<sub>2/3</sub>]O<sub>2</sub> electrode</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (f) the corresponding calculated lattice parameters for o-XRD pattern</strong><sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup><strong>; (g)from experimentally calculated data and data predicted by first-principles calculations</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup><strong>; (h) Contour maps in selected 2<i>θ</i> ranges obtained from In operando XRD patterns of P2-K<sub>0.75</sub>MNFO<sub>2</sub> during the charge-discharge process in the range of 1.5</strong>~<strong>3.9 V (K/K<sup>+</sup>)</strong><sup>[<xref ref-type="bibr" rid="R13">13</xref>]</sup>Fig. 33.1.3 改性研究
... [44]; (f) the corresponding calculated lattice parameters for o-XRD pattern[44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
... [44]; (g)from experimentally calculated data and data predicted by first-principles calculations[13]; (h) Contour maps in selected 2θ ranges obtained from In operando XRD patterns of P2-K0.75MNFO2 during the charge-discharge process in the range of 1.5~3.9 V (K/K+)[13]Fig. 33.1.3 改性研究
... 为了提高单过渡金属氧化物正极材料的电化学性能,在锰基体系中引入其他过渡金属元素设计二元过渡金属氧化物体系是一种行之有效的方法.该方法已很好地应用于锂离子电池和钠离子电池的层状氧化物正极中[40-42].通过过渡金属离子之间的协同作用,二元过渡金属氧化物可以优化锰基层状氧化物的晶格结构,从而提高其结构稳定性,进而表现出优异的综合电化学性能.二元过渡金属氧化物体系可以增加可逆容量,平滑电压分布,提高平均工作电压,并表现出理想的钾离子迁移率、优异的延展性、更丰富的氧化还原位点和更好的电化学活性[45-46].由于这些特性,一些二元过渡金属氧化物引起了钾离子电池系统研究人员越来越多的关注.近年来,研究人员发现对于锰基层状氧化物结构,采用金属离子替代层状K x TMO2的正极材料在连续的K+插入/提取过程中对提高结构稳定性也有很大帮助.如,Ti是层状氧化物材料中常见的非活性掺杂元素,可以提高材料的循环稳定性,稳定材料的晶体结构.尽管Mg/Mn基和Ti/Mn基二元过渡金属氧化物已被报道显示出良好的电化学性能,但Mg和Ti毕竟是电化学惰性元素,在材料中引入过多会降低理论容量.因此,这两种元素通常被少量地添加到层状氧化物中作为掺杂元素,以实现对电化学性能的有效调节.但对于动力电池来说,高的能量密度和循环稳定性都是重要的电化学性能指标,因此采用电化学活性元素取代的二元过渡金属氧化物体系可能是未来的研究重点,如在Ni/Mn基层状氧化物中,连续发生的多电子氧化还原反应,可在高电压范围内产生理想的能量密度.Jo等[44]报道了一种Ni取代的P2-K0.75[Ni1/3Mn2/3]O2样品,当截止电压设置为4.3 V时,其在20 mA/g的电流密度下拥有110 mAh/g的比容量,并且循环300圈后仍有86%的容量保持率.当脱钾(氧化)至4.3 V时,P2-K0.75[Ni1/3Mn2/3]O2的主衍射峰(002)和(004)的角度逐渐向低角度移动,而(100)、(102)、(103)和(104)衍射峰的角度逐渐向高角度移动[图3(e)],即K+从结构中脱嵌导致结构中氧离子之间的相互排斥作用诱发了a轴逐渐减小而尾轴逐渐增大[图3(f)].在充电过程中没有检测到其他相的产生.在嵌钾(还原)至1.5 V时,P2-K0.75[Ni1/3Mn2/3]O2的主峰可逆地回到了原来的布拉格峰位置,揭示了P2-K0.75[Ni1/3Mn2/3]O2的整体储钾机制为单相反应,充放电时晶格变化较小. ...
0
0
4
... 一般情况下,P3-K x MnO2正极材料在充放电过程中会发生多相转变,在脱钾状态下向O3型层状结构转变,从而引起较大的体积变化.这是由于钾位空位形成的氧-氧排斥作用的结果.这可能是容量衰减的原因,也同样导致了具有这种现象的样品在电化学测试时呈现出非平滑的充放电曲线.例如,Kim等[48]报道了P3-K0.5MnO2,这项工作可以帮助大家更好地理解层状P3型K0.5MnO2中的晶体结构演变,并且对钾离子电池新型正极材料的设计和开发提供了见解.图4(a)展示了P3-K0.5MnO2正极在2 mA/g电流密度下循环时收集的原位XRD图谱.我们观察到在特定K含量(K x MnO2中的0.395<x<0.425和0.316<x<0.364)与其他K组成的固溶态的两相反应.K脱嵌后,(003)和(006)峰位移到较低的角度[图4(b)、(c)],表明MnO2平板距离扩大.更重要的是,在K x MnO2中,(015)峰消失,在x≈0.41处出现一个新的(104)峰.这一特征表明随着钾离子的去除,从P3结构转变为O3结构[49].并且,当K x MnO2进一步脱嵌K至x≈0.34后,在(003)和(006)峰的左边出现了一组新的XRD峰,如图4(b)、(c)所示,表明发生了另一个相变.在1.5~4.0 V的工作电压窗口中,在电流密度为16 mA/g时,其初始比容量为140 mAh/g.即使在如此低的截止电压下测试,通过原位X射线(XRD)表征也观察到的P3-O3相变,这应该是循环性能下降的原因. ...
... [48];(g) P3-K0.75MnO2 和P3-K0.75[Co0.5Mn0.5]O2 的晶体结构示意图;(h) P3-K x [Co0.5Mn0.5]O2(0.25≤ x ≤0.75)的结构变化;(i) P3-K x [Co0.5Mn0.5]O2 的XRD谱图;(j) 通过DFT计算和XRD数据得到的P3-K x [Co0.5Mn0.5]O2 的晶格参数的对比图[57];(k) 花生状P3型K0.45Mn0.5Co0.5O2 微粒的合成示意图[59];(l)、(m) P3型K0.45Mn0.5Co0.5O2 的SEM图像[59];(n) P3型K0.45Mn0.5Co0.5O2 在300 mA/g电流密度下的长期循环性能[59];(o) K0.5MnO2 的循环伏安图<strong>(a) Typical charge/discharge profiles of P3-type K<sub>0.5</sub>MnO<sub>2</sub> at a current rate of 2 mA/g; (b)</strong>~<strong>(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K<sub>0.5</sub>MnO<sub>2</sub> at 18°</strong>~<strong>20°; (f) comparison with simulated XRD patterns of the O3 and P3structures</strong><sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup><strong>; (g) P3-K<sub>0.75</sub>MnO<sub>2</sub> and P3-K<sub>0.75</sub>[Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (h) Predicted structural change in P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> (0.25</strong>≤ <strong><i>x</i></strong> ≤<strong>0.75); (i) operando XRD patterns of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (j) comparison of c-lattice parameters of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> obtained by first-principles calculations and operando XRD data</strong><sup>[<xref ref-type="bibr" rid="R57">57</xref>]</sup><strong>; (k) Schematic illustration of the synthesis of peanut-like P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> microparticles</strong><sup>[<xref ref-type="bibr" rid="R59">59</xref>]</sup><strong>; (l), (m) SEM images of P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub>; (n) long-term cycling performance of K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s</strong>Fig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... [48]; (g) P3-K0.75MnO2 and P3-K0.75[Co0.5Mn0.5]O2; (h) Predicted structural change in P3-K x [Co0.5Mn0.5]O2 (0.25≤ x ≤0.75); (i) operando XRD patterns of P3-K x [Co0.5Mn0.5]O2; (j) comparison of c-lattice parameters of P3-K x [Co0.5Mn0.5]O2 obtained by first-principles calculations and operando XRD data[57]; (k) Schematic illustration of the synthesis of peanut-like P3-type K0.45Mn0.5Co0.5O2 microparticles[59]; (l), (m) SEM images of P3-type K0.45Mn0.5Co0.5O2; (n) long-term cycling performance of K0.45Mn0.5Co0.5O2 at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/sFig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... 研究人员还发现传统的样品制备方法,例如固相方法,通常会产生不规则形状的颗粒,其表面结构容易被破坏,并逐渐渗透到本体相中,导致库仑效率低,容量衰减快.而相较下结构工程由于其优异的结构稳定性,可以对抗连续嵌入/脱嵌钾离子引起的机械应力而得到了很好的设计研究.控制合成具有良好形貌的层状氧化物材料,既能提高结构稳定性,又能抑制副反应,有利于充分发挥层状锰基正极结构的K+存储潜力[58].最近,报道了P3-K0.45Mn0.5Co0.5O2[59].Co占据Mn位点,以抑制Mn3+的Jahn-Teller效应,并且独特的花生状结构[图4(k)~(m)]不仅大大缩短了K+的扩散距离,还缓解了K+连续嵌入/脱嵌引起的内部应变,从而改善了反应动力学和结构稳定性,提高了P3-K0.45Mn0.5Co0.5O2的循环稳定性和倍率性能.这为实现令人满意的钾离子电池晶格结构设计提供了新的策略.其在电流密度为300 mA/g的条件下,具有稳定的循环能力,在300次循环后保持了73.8%的高比容量[图4(n)],之后仍然保持中空特征和高结晶度,没有任何杂质,证实了其突出的结构稳定性.值得注意的是,上述P3型锰基层状氧化物的电压窗口都未超过4 V,这是因为降低截止电压对于稳定P3-K x MnO2的层状结构也有帮助.例如[48]在1.5~4.2 V (vs K/K+)扫描时,在约3.7 V和4.1 V(vs K/K+)处观察到两个强氧化峰,而对应的还原峰不太明显[图4(o)].相反,在1.5~3.9 V扫描时,氧化峰和还原峰匹配良好,这些结果表明,K的脱嵌和嵌入过程可能导致高电压区发生不可逆的结构变化.因此,降低截止电压对于提高P3-K x MnO2的层状结构在充放电过程中的可逆性具有积极影响. ...
1
... 一般情况下,P3-K x MnO2正极材料在充放电过程中会发生多相转变,在脱钾状态下向O3型层状结构转变,从而引起较大的体积变化.这是由于钾位空位形成的氧-氧排斥作用的结果.这可能是容量衰减的原因,也同样导致了具有这种现象的样品在电化学测试时呈现出非平滑的充放电曲线.例如,Kim等[48]报道了P3-K0.5MnO2,这项工作可以帮助大家更好地理解层状P3型K0.5MnO2中的晶体结构演变,并且对钾离子电池新型正极材料的设计和开发提供了见解.图4(a)展示了P3-K0.5MnO2正极在2 mA/g电流密度下循环时收集的原位XRD图谱.我们观察到在特定K含量(K x MnO2中的0.395<x<0.425和0.316<x<0.364)与其他K组成的固溶态的两相反应.K脱嵌后,(003)和(006)峰位移到较低的角度[图4(b)、(c)],表明MnO2平板距离扩大.更重要的是,在K x MnO2中,(015)峰消失,在x≈0.41处出现一个新的(104)峰.这一特征表明随着钾离子的去除,从P3结构转变为O3结构[49].并且,当K x MnO2进一步脱嵌K至x≈0.34后,在(003)和(006)峰的左边出现了一组新的XRD峰,如图4(b)、(c)所示,表明发生了另一个相变.在1.5~4.0 V的工作电压窗口中,在电流密度为16 mA/g时,其初始比容量为140 mAh/g.即使在如此低的截止电压下测试,通过原位X射线(XRD)表征也观察到的P3-O3相变,这应该是循环性能下降的原因. ...
1
... 此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
1
... 此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
1
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
1
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
1
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
1
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
1
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
3
... 一般情况下,P3-K x MnO2正极材料在充放电过程中会发生多相转变,在脱钾状态下向O3型层状结构转变,从而引起较大的体积变化.这是由于钾位空位形成的氧-氧排斥作用的结果.这可能是容量衰减的原因,也同样导致了具有这种现象的样品在电化学测试时呈现出非平滑的充放电曲线.例如,Kim等[48]报道了P3-K0.5MnO2,这项工作可以帮助大家更好地理解层状P3型K0.5MnO2中的晶体结构演变,并且对钾离子电池新型正极材料的设计和开发提供了见解.图4(a)展示了P3-K0.5MnO2正极在2 mA/g电流密度下循环时收集的原位XRD图谱.我们观察到在特定K含量(K x MnO2中的0.395<x<0.425和0.316<x<0.364)与其他K组成的固溶态的两相反应.K脱嵌后,(003)和(006)峰位移到较低的角度[图4(b)、(c)],表明MnO2平板距离扩大.更重要的是,在K x MnO2中,(015)峰消失,在x≈0.41处出现一个新的(104)峰.这一特征表明随着钾离子的去除,从P3结构转变为O3结构[49].并且,当K x MnO2进一步脱嵌K至x≈0.34后,在(003)和(006)峰的左边出现了一组新的XRD峰,如图4(b)、(c)所示,表明发生了另一个相变.在1.5~4.0 V的工作电压窗口中,在电流密度为16 mA/g时,其初始比容量为140 mAh/g.即使在如此低的截止电压下测试,通过原位X射线(XRD)表征也观察到的P3-O3相变,这应该是循环性能下降的原因.
<strong>(a) Typical charge/discharge profiles of P3-type K<sub>0.5</sub>MnO<sub>2</sub> at a current rate of 2 mA/g; (b)</strong>~<strong>(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K<sub>0.5</sub>MnO<sub>2</sub> at 18°</strong>~<strong>20°; (f) comparison with simulated XRD patterns of the O3 and P3structures</strong><sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup><strong>; (g) P3-K<sub>0.75</sub>MnO<sub>2</sub> and P3-K<sub>0.75</sub>[Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (h) Predicted structural change in P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> (0.25</strong>≤ <strong><i>x</i></strong> ≤<strong>0.75); (i) operando XRD patterns of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (j) comparison of c-lattice parameters of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> obtained by first-principles calculations and operando XRD data</strong><sup>[<xref ref-type="bibr" rid="R57">57</xref>]</sup><strong>; (k) Schematic illustration of the synthesis of peanut-like P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> microparticles</strong><sup>[<xref ref-type="bibr" rid="R59">59</xref>]</sup><strong>; (l), (m) SEM images of P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub>; (n) long-term cycling performance of K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s</strong>Fig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... [57]; (k) Schematic illustration of the synthesis of peanut-like P3-type K0.45Mn0.5Co0.5O2 microparticles[59]; (l), (m) SEM images of P3-type K0.45Mn0.5Co0.5O2; (n) long-term cycling performance of K0.45Mn0.5Co0.5O2 at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/sFig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... 与P2相材料类似,发现P3相的Fe[52]、Ni[53]、Co[54]、Mg[55]等金属离子掺杂或取代与K+插入/提取过程中层状框架的稳定性增强有关,提高了循环稳定性.如Ramasamy等[56-57]研究了Co部分取代Mn对此类正极材料电化学性能的影响.由于晶体结构中Co3+取代了Mn3+,导致了电压分布的平稳变化,这不仅缓解了K+/空位排序对电压平台的影响,还可能缓解Mn3+的Jahn-Teller(JT)畸变诱发的层状结构变化.利用密度泛函理论(DFT)进行第一性原理计算得到P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2(x=0.75和0.25)的晶体结构,比较了P3-K x MnO2和P3-K x [Co0.5Mn0.5]O2在Mn3+作用下的Jahn-Teller畸变.除了原始P3-K0.75MnO2结构中MnO6八面体上的两种Mn-O键拉长外,Co3+取代的P3-K0.75[Co0.5Mn0.5]O2结构中MnO6八面体的所有Mn—O键长度都相似[图4(g)].这表明Co3+取代可以有效缓解结构中Mn3+引起的JT畸变.这种改善可能会使得P3-K x [Co0.5Mn0.5]O2平稳电压分布.预计在八面体环境中,Mn4+/3+氧化还原对的较少利用以及与CoO6八面体(如O-Mn-O-Co-O)共享氧将使得JT畸变最小化,提高循环时的结构稳定性[图4(h)].基于X射线衍射和第一性原理计算的联合研究[图4(i)、(j)],证实过渡金属层之间Mn和Co的共存可以调节相变的程度进而有效地增强了层状稳定性,从而为长循环寿命和优异的倍率性能提供了支持.实验结果表明,P3-K0.54[Co0.5Mn0.5]O2正极具有120.4 mAh/g的高可逆比容量,平均工作电压为2.85 V(vs. K+/K).在20 mA/g下超过100次循环,可保持其初始容量的85%.即使在500 mA/g的循环测试下,也显示出78 mAh/g的出色电化学性能. ...
1
... 研究人员还发现传统的样品制备方法,例如固相方法,通常会产生不规则形状的颗粒,其表面结构容易被破坏,并逐渐渗透到本体相中,导致库仑效率低,容量衰减快.而相较下结构工程由于其优异的结构稳定性,可以对抗连续嵌入/脱嵌钾离子引起的机械应力而得到了很好的设计研究.控制合成具有良好形貌的层状氧化物材料,既能提高结构稳定性,又能抑制副反应,有利于充分发挥层状锰基正极结构的K+存储潜力[58].最近,报道了P3-K0.45Mn0.5Co0.5O2[59].Co占据Mn位点,以抑制Mn3+的Jahn-Teller效应,并且独特的花生状结构[图4(k)~(m)]不仅大大缩短了K+的扩散距离,还缓解了K+连续嵌入/脱嵌引起的内部应变,从而改善了反应动力学和结构稳定性,提高了P3-K0.45Mn0.5Co0.5O2的循环稳定性和倍率性能.这为实现令人满意的钾离子电池晶格结构设计提供了新的策略.其在电流密度为300 mA/g的条件下,具有稳定的循环能力,在300次循环后保持了73.8%的高比容量[图4(n)],之后仍然保持中空特征和高结晶度,没有任何杂质,证实了其突出的结构稳定性.值得注意的是,上述P3型锰基层状氧化物的电压窗口都未超过4 V,这是因为降低截止电压对于稳定P3-K x MnO2的层状结构也有帮助.例如[48]在1.5~4.2 V (vs K/K+)扫描时,在约3.7 V和4.1 V(vs K/K+)处观察到两个强氧化峰,而对应的还原峰不太明显[图4(o)].相反,在1.5~3.9 V扫描时,氧化峰和还原峰匹配良好,这些结果表明,K的脱嵌和嵌入过程可能导致高电压区发生不可逆的结构变化.因此,降低截止电压对于提高P3-K x MnO2的层状结构在充放电过程中的可逆性具有积极影响. ...
5
... 一般情况下,P3-K x MnO2正极材料在充放电过程中会发生多相转变,在脱钾状态下向O3型层状结构转变,从而引起较大的体积变化.这是由于钾位空位形成的氧-氧排斥作用的结果.这可能是容量衰减的原因,也同样导致了具有这种现象的样品在电化学测试时呈现出非平滑的充放电曲线.例如,Kim等[48]报道了P3-K0.5MnO2,这项工作可以帮助大家更好地理解层状P3型K0.5MnO2中的晶体结构演变,并且对钾离子电池新型正极材料的设计和开发提供了见解.图4(a)展示了P3-K0.5MnO2正极在2 mA/g电流密度下循环时收集的原位XRD图谱.我们观察到在特定K含量(K x MnO2中的0.395<x<0.425和0.316<x<0.364)与其他K组成的固溶态的两相反应.K脱嵌后,(003)和(006)峰位移到较低的角度[图4(b)、(c)],表明MnO2平板距离扩大.更重要的是,在K x MnO2中,(015)峰消失,在x≈0.41处出现一个新的(104)峰.这一特征表明随着钾离子的去除,从P3结构转变为O3结构[49].并且,当K x MnO2进一步脱嵌K至x≈0.34后,在(003)和(006)峰的左边出现了一组新的XRD峰,如图4(b)、(c)所示,表明发生了另一个相变.在1.5~4.0 V的工作电压窗口中,在电流密度为16 mA/g时,其初始比容量为140 mAh/g.即使在如此低的截止电压下测试,通过原位X射线(XRD)表征也观察到的P3-O3相变,这应该是循环性能下降的原因.
<strong>(a) Typical charge/discharge profiles of P3-type K<sub>0.5</sub>MnO<sub>2</sub> at a current rate of 2 mA/g; (b)</strong>~<strong>(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K<sub>0.5</sub>MnO<sub>2</sub> at 18°</strong>~<strong>20°; (f) comparison with simulated XRD patterns of the O3 and P3structures</strong><sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup><strong>; (g) P3-K<sub>0.75</sub>MnO<sub>2</sub> and P3-K<sub>0.75</sub>[Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (h) Predicted structural change in P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> (0.25</strong>≤ <strong><i>x</i></strong> ≤<strong>0.75); (i) operando XRD patterns of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (j) comparison of c-lattice parameters of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> obtained by first-principles calculations and operando XRD data</strong><sup>[<xref ref-type="bibr" rid="R57">57</xref>]</sup><strong>; (k) Schematic illustration of the synthesis of peanut-like P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> microparticles</strong><sup>[<xref ref-type="bibr" rid="R59">59</xref>]</sup><strong>; (l), (m) SEM images of P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub>; (n) long-term cycling performance of K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s</strong>Fig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... [59];(n) P3型K0.45Mn0.5Co0.5O2 在300 mA/g电流密度下的长期循环性能[59];(o) K0.5MnO2 的循环伏安图<strong>(a) Typical charge/discharge profiles of P3-type K<sub>0.5</sub>MnO<sub>2</sub> at a current rate of 2 mA/g; (b)</strong>~<strong>(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K<sub>0.5</sub>MnO<sub>2</sub> at 18°</strong>~<strong>20°; (f) comparison with simulated XRD patterns of the O3 and P3structures</strong><sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup><strong>; (g) P3-K<sub>0.75</sub>MnO<sub>2</sub> and P3-K<sub>0.75</sub>[Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (h) Predicted structural change in P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> (0.25</strong>≤ <strong><i>x</i></strong> ≤<strong>0.75); (i) operando XRD patterns of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (j) comparison of c-lattice parameters of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> obtained by first-principles calculations and operando XRD data</strong><sup>[<xref ref-type="bibr" rid="R57">57</xref>]</sup><strong>; (k) Schematic illustration of the synthesis of peanut-like P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> microparticles</strong><sup>[<xref ref-type="bibr" rid="R59">59</xref>]</sup><strong>; (l), (m) SEM images of P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub>; (n) long-term cycling performance of K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s</strong>Fig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... [59];(o) K0.5MnO2 的循环伏安图<strong>(a) Typical charge/discharge profiles of P3-type K<sub>0.5</sub>MnO<sub>2</sub> at a current rate of 2 mA/g; (b)</strong>~<strong>(d) In situ XRD pattern taken for 2 h scanning rate per pattern; (e) XRD peak comparison of as-prepared (scan #1), scan #7, and scan #14 P3-K<sub>0.5</sub>MnO<sub>2</sub> at 18°</strong>~<strong>20°; (f) comparison with simulated XRD patterns of the O3 and P3structures</strong><sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup><strong>; (g) P3-K<sub>0.75</sub>MnO<sub>2</sub> and P3-K<sub>0.75</sub>[Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (h) Predicted structural change in P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> (0.25</strong>≤ <strong><i>x</i></strong> ≤<strong>0.75); (i) operando XRD patterns of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub>; (j) comparison of c-lattice parameters of P3-K <i><sub>x</sub></i> [Co<sub>0.5</sub>Mn<sub>0.5</sub>]O<sub>2</sub> obtained by first-principles calculations and operando XRD data</strong><sup>[<xref ref-type="bibr" rid="R57">57</xref>]</sup><strong>; (k) Schematic illustration of the synthesis of peanut-like P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> microparticles</strong><sup>[<xref ref-type="bibr" rid="R59">59</xref>]</sup><strong>; (l), (m) SEM images of P3-type K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub>; (n) long-term cycling performance of K<sub>0.45</sub>Mn<sub>0.5</sub>Co<sub>0.5</sub>O<sub>2</sub> at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/s</strong>Fig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... [59]; (l), (m) SEM images of P3-type K0.45Mn0.5Co0.5O2; (n) long-term cycling performance of K0.45Mn0.5Co0.5O2 at 300 mA/g; (o) Cyclic voltammograms at a scan rate of 0.03 mV/sFig. 4
此外,在锰基电极材料的应用中还面临其他的挑战应该得到解决,如Mn3+的Jahn-Teller畸变的存在,它诱发了在脱钾过程(放电状态下)中八面体环境中的Mn-O在一个方向上的延伸.据报道,Mn3+的Jahn-Teller畸变导致MnO6八面体中部分Mn-O键长度与其他Mn-O键长度不同,这是由于Mn离子在d轨道上的电子构型[50].这不仅导致了层状结构的紊乱,也可能是P3-K x MnO2在电化学测试时显示不平滑的充放电曲线的原因.这种现象在钠离子电池体系中也经常被观测到,例如Na x MnO2中,Mn3+的Jahn-Teller畸变破坏了Na x MnO2的结构稳定性,形成特定的钠空位,这导致Na x MnO2在充放电过程中出现多个电压阶跃[51].因此,必须克服这些挑战,减轻脱钾过程中的结构转变,避免Mn3+在晶体结构中的Jahn-Teller效应引起的结构紊乱. ...
... 研究人员还发现传统的样品制备方法,例如固相方法,通常会产生不规则形状的颗粒,其表面结构容易被破坏,并逐渐渗透到本体相中,导致库仑效率低,容量衰减快.而相较下结构工程由于其优异的结构稳定性,可以对抗连续嵌入/脱嵌钾离子引起的机械应力而得到了很好的设计研究.控制合成具有良好形貌的层状氧化物材料,既能提高结构稳定性,又能抑制副反应,有利于充分发挥层状锰基正极结构的K+存储潜力[58].最近,报道了P3-K0.45Mn0.5Co0.5O2[59].Co占据Mn位点,以抑制Mn3+的Jahn-Teller效应,并且独特的花生状结构[图4(k)~(m)]不仅大大缩短了K+的扩散距离,还缓解了K+连续嵌入/脱嵌引起的内部应变,从而改善了反应动力学和结构稳定性,提高了P3-K0.45Mn0.5Co0.5O2的循环稳定性和倍率性能.这为实现令人满意的钾离子电池晶格结构设计提供了新的策略.其在电流密度为300 mA/g的条件下,具有稳定的循环能力,在300次循环后保持了73.8%的高比容量[图4(n)],之后仍然保持中空特征和高结晶度,没有任何杂质,证实了其突出的结构稳定性.值得注意的是,上述P3型锰基层状氧化物的电压窗口都未超过4 V,这是因为降低截止电压对于稳定P3-K x MnO2的层状结构也有帮助.例如[48]在1.5~4.2 V (vs K/K+)扫描时,在约3.7 V和4.1 V(vs K/K+)处观察到两个强氧化峰,而对应的还原峰不太明显[图4(o)].相反,在1.5~3.9 V扫描时,氧化峰和还原峰匹配良好,这些结果表明,K的脱嵌和嵌入过程可能导致高电压区发生不可逆的结构变化.因此,降低截止电压对于提高P3-K x MnO2的层状结构在充放电过程中的可逆性具有积极影响. ...
... [61]; (b) Visualization of Jahn-Teller distortion[61]; (c) Long-term cycling performance of K0.5Mn0.6Co0.2Fe0.1Mg0.1O2 at 1 A/g[61]; (e) In situ XRD patterns of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (f) HRTEM images of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [61]; (c) Long-term cycling performance of K0.5Mn0.6Co0.2Fe0.1Mg0.1O2 at 1 A/g[61]; (e) In situ XRD patterns of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (f) HRTEM images of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [61]; (e) In situ XRD patterns of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (f) HRTEM images of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
(a) In situ XRD characterization of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (b) Visualization of Jahn-Teller distortion<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (c) Long-term cycling performance of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2 </sub>at 1 A/g<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (e) In situ XRD patterns of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (f) HRTEM images of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (g) Structural changes in K <i><sub>x</sub></i> Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> during K<sup>+</sup> insertion<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (h) Cycling performance of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> and KMO at 500 mA/g<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66];(g) K0.35Mn0.8Fe0.1Cu0.1O2 在K+ 嵌入过程中的结构变化示意图[66];(h) K0.35Mn0.8Fe0.1Cu0.1O2 在500 mA/g时的循环性能[66](a) In situ XRD characterization of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (b) Visualization of Jahn-Teller distortion<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (c) Long-term cycling performance of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2 </sub>at 1 A/g<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (e) In situ XRD patterns of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (f) HRTEM images of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (g) Structural changes in K <i><sub>x</sub></i> Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> during K<sup>+</sup> insertion<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (h) Cycling performance of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> and KMO at 500 mA/g<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66];(h) K0.35Mn0.8Fe0.1Cu0.1O2 在500 mA/g时的循环性能[66](a) In situ XRD characterization of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (b) Visualization of Jahn-Teller distortion<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (c) Long-term cycling performance of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2 </sub>at 1 A/g<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (e) In situ XRD patterns of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (f) HRTEM images of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (g) Structural changes in K <i><sub>x</sub></i> Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> during K<sup>+</sup> insertion<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (h) Cycling performance of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> and KMO at 500 mA/g<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66](a) In situ XRD characterization of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (b) Visualization of Jahn-Teller distortion<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (c) Long-term cycling performance of K<sub>0.5</sub>Mn<sub>0.6</sub>Co<sub>0.2</sub>Fe<sub>0.1</sub>Mg<sub>0.1</sub>O<sub>2 </sub>at 1 A/g<sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup>; (e) In situ XRD patterns of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (f) HRTEM images of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub><sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (g) Structural changes in K <i><sub>x</sub></i> Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> during K<sup>+</sup> insertion<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>; (h) Cycling performance of K<sub>0.35</sub>Mn<sub>0.8</sub>Fe<sub>0.1</sub>Cu<sub>0.1</sub>O<sub>2</sub> and KMO at 500 mA/g<sup>[<xref ref-type="bibr" rid="R66">66</xref>]</sup>Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66]; (f) HRTEM images of K0.35Mn0.8Fe0.1Cu0.1O2[66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66]; (g) Structural changes in K x Mn0.8Fe0.1Cu0.1O2 during K+ insertion[66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66]; (h) Cycling performance of K0.35Mn0.8Fe0.1Cu0.1O2 and KMO at 500 mA/g[66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [66]Fig. 5
事实上,降低截止电压是以降低可逆容量和能量密度为代价来实现稳定循环性能的,并不是理想策略.因此,选择合适的晶格结构设计策略,以提高其在高压区域的结构稳定性成为迫切需要解决的问题.在P2型K x MnO2中,报道了P2-K0.6Mn0.8Ni0.1Ti0.1O2[64],已经证实含Ti的多元过渡金属氧化物可以在高工作电压下显示出良好的电化学性能,而这对于P3型层状结构同样是可行之策[图5(d)].设计了一种在4.2 V高截止电压下工作的正极材料P3-K0.4Fe0.1Mn0.8Ti0.1O2[65],该材料具有良好的储钾性能.在20 mA/g时具有117 mAh/g的可逆钾存储容量.原位X射线衍射揭示了K+插入/脱出时的转变,体积变化为0.5%,确保了超过300个周期的长周期稳定性.在前两次充放电过程中,仅观察到有规律的峰移,表明固溶反应具有适度的结构演化.随着K+从主体结构中脱插,(003)和(006)衍射峰不断向较低的角度移动,而(101)和(012)衍射峰则向较高的角度移动.峰位的逐渐变化归因于结构沿c轴方向的膨胀.Mn3+→Mn4+和Fe3+→Fe4+在放电时观察到可逆现象,表明电极材料具有高度的结构可逆性.P3型化合物在充电时通常经历P3到O3的转变,导致K+迁移率缓慢和容量快速退化.这可以归因于O相框架内较高的活化势垒以及晶体结构的显著收缩.在这个实验中,K0.4Fe0.1Mn0.8Ti0.1O2经历了固溶体反应,没有明显的P3-O3转变,这可能是该材料具有良好的电化学性能的原因. ...
... [69]<strong>(a) Schematic diagram of the effect of Rb and Mg substitution on the crystal structure; (b) The stacking of P3-type and O3-type layered structures along the <i>c</i>-axis direction of the TM slabs; (c) Representative HRTEM images of the K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> powder and K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> test cathode placed in the battery environment without cycling; (d) The voltage curves and representative corresponding in situ XRD patterns of K<sub>0.45</sub>MnO<sub>2</sub> and K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> at 70 mA/g in the range of 1.5</strong>—<strong>3.9 V.; (e) The <i>c</i>-lattice parameter of K<sub>0.45</sub>MnO<sub>2</sub> and K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> calculated from in situ XRD patterns; (f) Charge GITT result of K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> at 20 mA/g, and the corresponding <i>D</i><sub>K+</sub>; (g) Cycling performance of K<sub>0.45</sub>Rb<sub>0.05</sub>Mn<sub>0.85</sub>Mg<sub>0.15</sub>O<sub>2</sub> at 200 mA/g</strong><sup>[<xref ref-type="bibr" rid="R69">69</xref>]</sup>Fig. 64 总结与展望



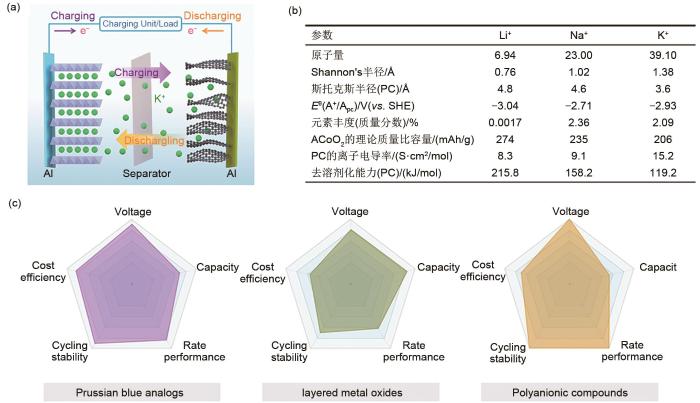
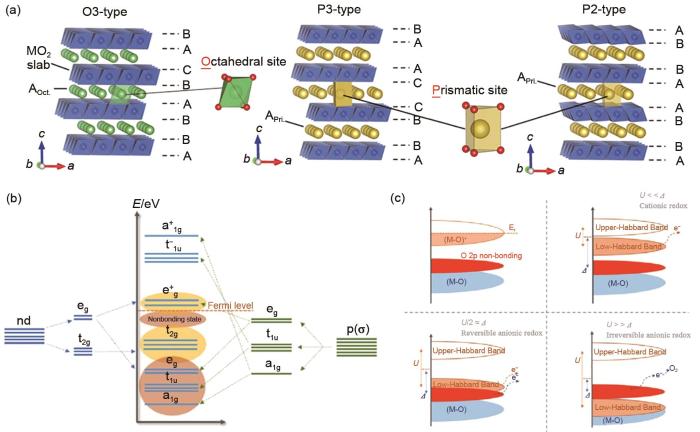
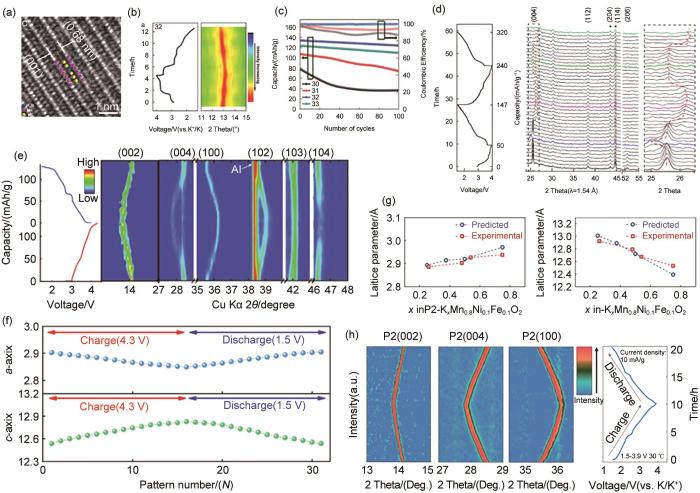
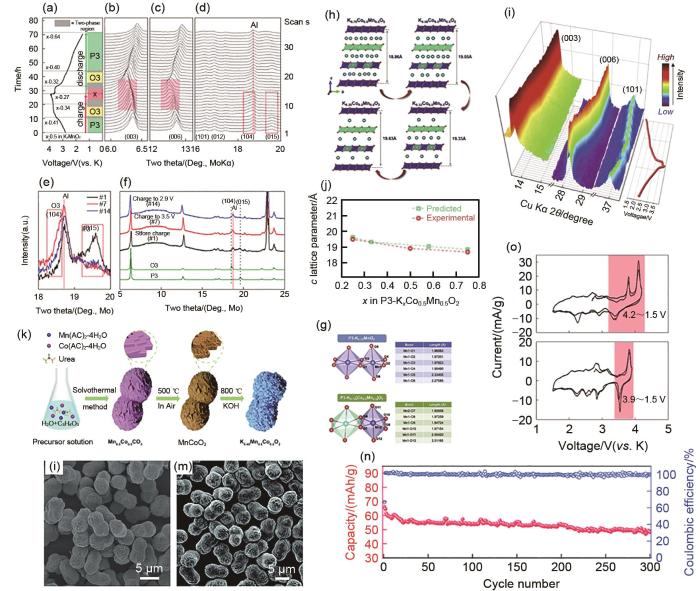

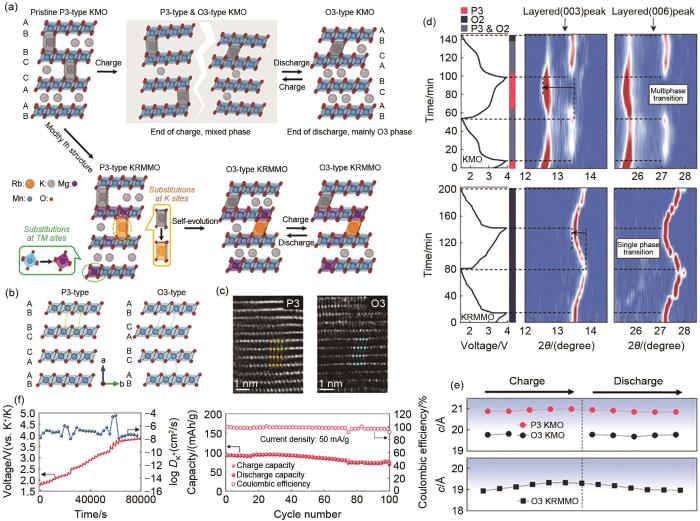

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}