Research progress of hard carbon anode materials for sodium ion batteries
LIU Fei,1,2,3, ZHAO Peiwen1,2,3, ZHAO Jingxiang1,2,3, SUN Xianwei1,2,3, LI Miaomiao1,2,3, WANG Jinghao1,2,3, YIN Yanxin1,2,3, DAI Zuoqiang,1,2,3, ZHENG Lili1,2,3
1.College of Mechanical and Electrical Engineering, Qingdao University
2.Engineering Technology Center of Power Integration and Energy Storage System
3.National and Local Joint Engineering Technology Center for Intelligent Power Integration Technology of Electric Vehicles (Qingdao), Qingdao 266071, Shandong, China
With the development of high-performance electrode materials and the study of the mechanism, the electrochemical performance of sodium-ion batteries has been greatly improved. Hard carbon has become recognized as the most mature and commercialized anode material. However, it still faces problems such as low initial coulomb efficiency and poor rate capability. At the same time, great efforts have been devoted to in-depth research on the mechanism of sodium storage in hard carbons, and to explore synthetic methods to improve performance and reduce costs. However, there are still disagreements on the sodium storage mechanism, especially the sodium storage mechanism in the plateau region. Through the study of recent literature, based on the three different sodium storage processes of hard carbon material intercalation, adsorption and nanopore filling, the "intercalation-adsorption", "adsorption-intercalation" and other various forms of composite sodium storage mechanisms are emphatically introduced. Then, the effects of specific surface area, pores, defects, interlayer spacing and functional groups on the rate capability and initial Coulomb efficiency of hard carbon anode materials were analyzed based on the in-depth understanding of the sodium storage mechanism of hard carbon materials. At the same time, the effects of structure optimization and surface modification of coating method on improving the rate performance and initial coulombic efficiency of hard carbon anode materials are introduced. In order to promote the practical application of hard carbon, the effect of electrolyte optimization on improve the performance of ICE and rate capability of hard carbon is expounded. Comprehensive analysis shows that hard carbon material modification and electrolyte optimization are promising to achieve high rate capability, high initial coulombic efficiency and cycle stability at the same time.
Keywords:sodium-ion battery
;
hard carbon
;
anode material
;
initial coulombic efficiency
;
rate capability
;
sodium storage mechanism
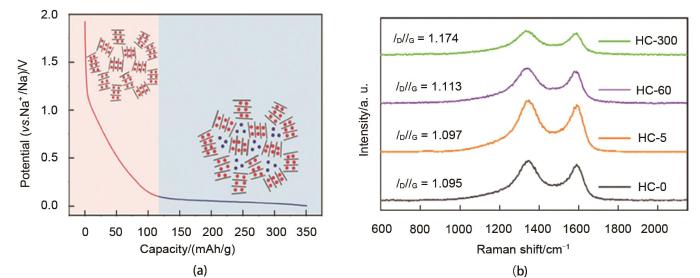
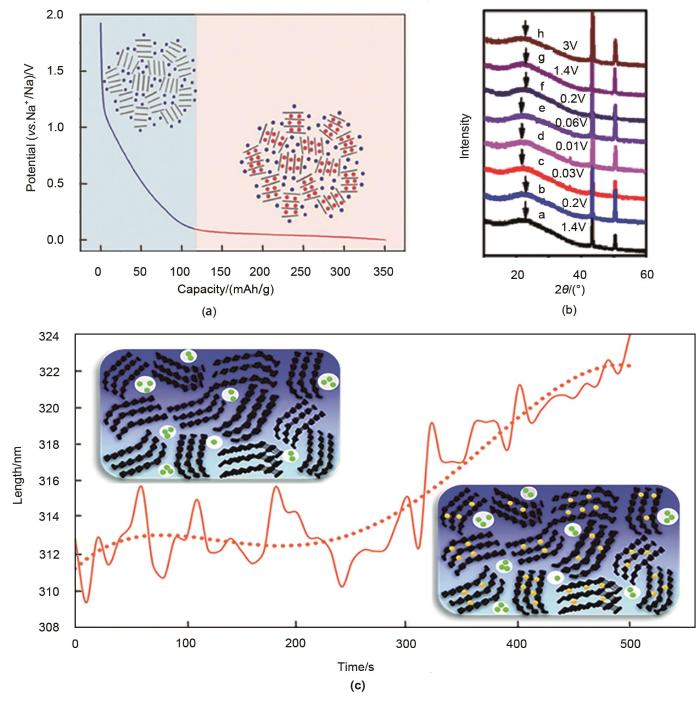
Fig. 2
(a) "Adsorption-intercalation" mechanism[8]; (b) XRD at different potentials[10]; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation[11]
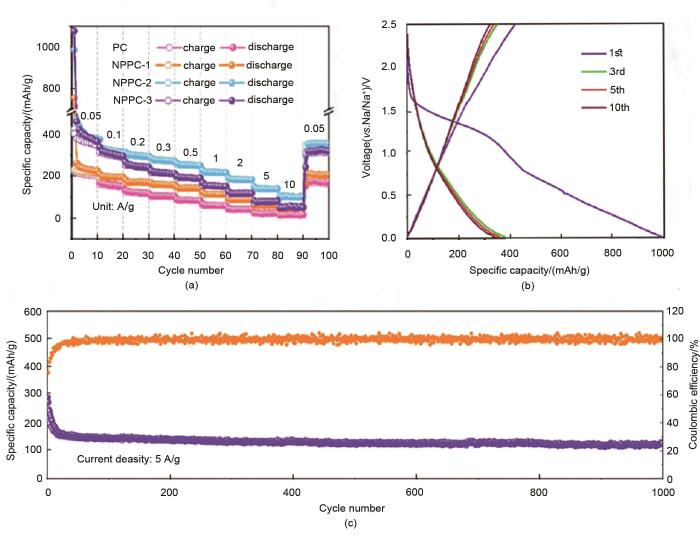
Fig. 3
Sodium storage capacity of NPPC and PC (a) Rate capability; (b) Charge-discharge curve of NPPC-2 at 50 mA/g; (c) Long-cycle performance of NPPC-2 at 5 A/g[25]
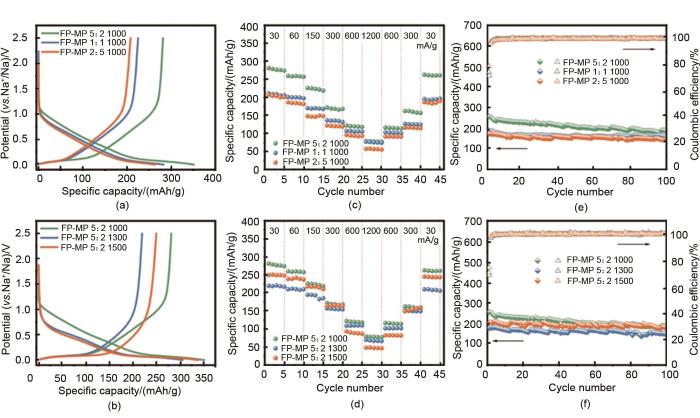
Fig. 4
(a), (b) Initial galvanostatic discharge/charge profiles of all the resulting hard-soft carbon composites at 30 mA/g; (c), (d) The rate capability of all the resulting hard-soft carbon composites; (e), (f) The cycling performance of all the resulting hard-soft carbon composites at 150 mA/g[35]
Fig. 6
(a) Initial charge-discharge profiles of hard carbon electrodes coated with different cycles of Al2O3 at a current density of 20 mA/g; (b) Cycling performance of hard carbon electrodes coated with different cycles of Al2O3 at 50 mA/g(The current rate is 20 mA/g in the initial five cycles); (c) Cycling performance of SHC-ALD0 and SHC-ALD20 at a current rate of 50 mA/g(The current rate is 20 mA/g in the initial five cycles)[37]
Fig. 7
(a) Cycling performance of HC at different current densities in TEGDME[40]; (b) Cycling performance of HC at different current densities in carbonate[40]; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging[41]; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging[41]; (e) Charge-discharge curves of hard carbon in different electrolytes[51]; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g[51]
VAALMA C, BUCHHOLZ D, WEIL M, et al. A cost and resource analysis of sodium-ion batteries[J]. Nature Reviews Materials, 2018, 3: doi: 10.1038/natrevmats.2018.13.
LIU Y Y, MERINOV B V, GODDARD W A 3rd. Origin of low sodium capacity in graphite and generally weak substrate binding of Na and Mg among alkali and alkaline earth metals[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(14): 3735-3739.
LI C H, SUN Y, WU Q J, et al. A novel design strategy of a practical carbon anode material from a single lignin-based surfactant source for sodium-ion batteries[J]. Chemical Communications (Cambridge, England), 2020, 56(45): 6078-6081.
LI Y, YUAN Y F, BAI Y, et al. Insights into the Na+ storage mechanism of phosphorus-functionalized hard carbon as ultrahigh capacity anodes[J]. Advanced Energy Materials, 2018, 8(18): doi: 10.1002/aenm.201702781.
STEVENS D A, DAHN J R. High capacity anode materials for rechargeable sodium-ion batteries[J]. Journal of the Electrochemical Society, 2000, 147(4): 1271.
STEVENS D A, DAHN J R. The mechanisms of lithium and sodium insertion in carbon materials[J]. Journal of the Electrochemical Society, 2001, 148(8): doi: 10.1149/1.1379565.
ILIC I K, SCHUTJAJEW K, ZHANG W Y, et al. Changes of porosity of hard carbons during mechanical treatment and the relevance for sodium-ion anodes[J]. Carbon, 2022, 186: 55-63.
QIU S, XIAO L F, SUSHKO M L, et al. Manipulating adsorption-insertion mechanisms in nanostructured carbon materials for high-efficiency sodium ion storage[J]. Advanced Energy Materials, 2017, 7(17): doi: 10.1002/aenm.201700403.
CAO Y L, XIAO L F, SUSHKO M L, et al. Sodium ion insertion in hollow carbon nanowires for battery applications[J]. Nano Letters, 2012, 12(7): 3783-3787.
JIN Q Z. The study on sodium storage mechanisms and structure-activity relationships of hard carbon electrode[D]. Wuhan: Huazhong University of Science and Technology, 2020.
WANG K, XU Y B, LI Y, et al. Sodium storage in hard carbon with curved graphene platelets as the basic structural units[J]. Journal of Materials Chemistry A, 2019, 7(7): 3327-3335.
ALVIN S, CAHYADI H S, HWANG J, et al. Revealing the intercalation mechanisms of lithium, sodium, and potassium in hard carbon[J]. Advanced Energy Materials, 2020, 10(20): doi: 10.1002/aenm.202000283.
CHEN X Y, FANG Y L, TIAN J Y, et al. Electrochemical insight into the sodium-ion storage mechanism on a hard carbon anode[J]. ACS Applied Materials & Interfaces, 2021, 13(16): 18914-18922.
AU H, ALPTEKIN H, JENSEN A C S, et al. A revised mechanistic model for sodium insertion in hard carbons[J]. Energy & Environmental Science, 2020, 13(10): 3469-3479.
CAI C C, CHEN Y A, HU P, et al. Regulating the interlayer spacings of hard carbon nanofibers enables enhanced pore filling sodium storage[J]. Small, 2022, 18(6): doi: 10.1002/smll.202105303.
CHEN X Y, TIAN J Y, LI P, et al. An overall understanding of sodium storage behaviors in hard carbons by an "adsorption-intercalation/filling" hybrid mechanism[J]. Advanced Energy Materials, 2022, 12(24): doi: 10.1002/aenm.202200886.
JIANG N, CHEN L, JIANG H, et al. Introducing the solvent co-intercalation mechanism for hard carbon with ultrafast sodium storage[J]. Small, 2022, 18(15): doi: 10.1002/smll.202108092.
ESCHER I, A FERRERO G, GOKTAS M, et al. In situ (operando) electrochemical dilatometry as a method to distinguish charge storage mechanisms and metal plating processes for sodium and lithium ions in hard carbon battery electrodes[J]. Advanced Materials Interfaces, 2022, 9(8): doi: 10.1002/admi.202100596.
CHEN J, HU T, ZOU Z, et al. Pre-doping iodine to restrain formation of low-active graphitic-N in hard carbon for significantly boosting sodium storage performance[J]. Carbon, 2022, 186: 193-204.
SENTHIL C, PARK J W, SHAJI N, et al. Biomass seaweed-derived nitrogen self-doped porous carbon anodes for sodium-ion batteries: Insights into the structure and electrochemical activity[J]. Journal of Energy Chemistry, 2022, 64: 286-295.
SUN Y, WU Q J, WANG Y D, et al. Protein-derived 3D amorphous carbon with N, O doping as high rate and long lifespan anode for potassium ion batteries[J]. Journal of Power Sources, 2021, 512: doi: 10.1016/j.jpowsour.2021.230530.
PEI Z X, MENG Q Q, WEI L, et al. Toward efficient and high rate sodium-ion storage: A new insight from dopant-defect interplay in textured carbon anode materials[J]. Energy Storage Materials, 2020, 28: 55-63.
FAN L L, ZHANG X, FAN L P, et al. Boosting the high capacitance-controlled capacity of hard carbon by using surface oxygen functional groups for fast and stable sodium storage[J]. ACS Applied Energy Materials, 2021, 4(10): 11436-11446.
XING C, YANG D H, ZHANG Y, et al. Semi-closed synthesis of nitrogen and oxygen Co-doped mesoporous carbon for selective aqueous oxidation[J]. Green Energy & Environment, 2022, 7(1): 43-52.
CHEN C, HUANG Y, MENG Z Y, et al. Experimental design and theoretical evaluation of nitrogen and phosphorus dual-doped hierarchical porous carbon for high-performance sodium-ion storage[J]. Journal of Materials Science & Technology, 2021, 76: 11-19.
WU Z R, ZOU J, ZHANG Y, et al. Lignin-derived hard carbon anode for potassium-ion batteries: Interplay among lignin molecular weight, material structures, and storage mechanisms[J]. Chemical Engineering Journal, 2022, 427: doi: 10.1016/j.cej.2021.131547.
VELDEVI T, RAGHU S, KALAIVANI R A, et al. Waste tire derived carbon as potential anode for lithium-ion batteries[J]. Chemosphere, 2022, 288: doi: 10.1016/j.chemosphere.2021.132438.
YIN X P, ZHAO Y F, WANG X, et al. Modulating the graphitic domains of hard carbons derived from mixed pitch and resin to achieve high rate and stable sodium storage[J]. Small, 2022, 18(5): doi: 10.1002/smll.202105568.
TONNOIR H, HUO D, CANEVESI R L S, et al. Tannin-based hard carbons as high-performance anode materials for sodium-ion batteries[J]. Materials Today Chemistry, 2022, 23: doi: 10.1016/j.mtchem.2021.100614.
LI Y Q, LU Y X, MENG Q S, et al. Regulating pore structure of hierarchical porous waste cork-derived hard carbon anode for enhanced Na storage performance[J]. Advanced Energy Materials, 2019, 9(48): doi: 10.1002/aenm.201902852.
LI Y M, WU F F, XIONG S L. Embedding ZnSe nanoparticles in a porous nitrogen-doped carbon framework for efficient sodium storage[J]. Electrochimica Acta, 2019, 296: 582-589.
YOUN Y, GAO B, KAMIYAMA A, et al. Nanometer-size Na cluster formation in micropore of hard carbon as origin of higher-capacity Na-ion battery[J]. Npj Computational Materials, 2021, 7: 48.
KAMIYAMA A, KUBOTA K, IGARASHI D, et al. MgO-template synthesis of extremely high capacity hard carbon for Na-ion battery[J]. Angewandte Chemie, 2021, 133(10): 5174-5180.
HE X X, ZHAO J H, LAI W H, et al. Soft-carbon-coated, free-standing, low-defect, hard-carbon anode to achieve a 94% initial coulombic efficiency for sodium-ion batteries[J]. ACS Applied Materials & Interfaces, 2021, 13(37): 44358-44368.
LU H Y, CHEN X Y, JIA Y L, et al. Engineering Al2O3 atomic layer deposition: Enhanced hard carbon-electrolyte interface towards practical sodium ion batteries[J]. Nano Energy, 2019, 64: doi: 10.1016/j.nanoen.2019.103903.
WANG C C, SU W L. Ultrathin artificial solid electrolyte interface layer-coated biomass-derived hard carbon as an anode for sodium-ion batteries[J]. ACS Applied Energy Materials, 2022, 5(1): 1052-1064.
XIAO B W, SOTO F A, GU M, et al. Lithium-pretreated hard carbon as high-performance sodium-ion battery anodes[J]. Advanced Energy Materials, 2018, 8(24): doi: 10.1002/aenm.201801441.
HIRSH H S, SAYAHPOUR B, SHEN A, et al. Role of electrolyte in stabilizing hard carbon as an anode for rechargeable sodium-ion batteries with long cycle life[J]. Energy Storage Materials, 2021, 42: 78-87.
DONG R Q, ZHENG L M, BAI Y, et al. Elucidating the mechanism of fast Na storage kinetics in ether electrolytes for hard carbon anodes[J]. Advanced Materials, 2021, 33(36): doi: 10.1002/adma.202008810.
LI Y W, CHEN S M, XU S Y, et al. Impact of electrolyte salts on Na storage performance for high-surface-area carbon anodes[J]. ACS Applied Materials & Interfaces, 2021, 13(41): 48745-48752.
ZHANG J, WANG D W, LV W, et al. Achieving superb sodium storage performance on carbon anodes through an ether-derived solid electrolyte interphase [J]. Energy Environ Sci, 2017, 10(1): 370-376.
CHEN Y, ZHAO S, YU Y Y, et al. A general synthesis of mesoporous hollow carbon spheres with extraordinary sodium storage kinetics by engineering solvation structure[J]. Small, 2022, 18(10): doi: 10.1002/smll.202106513.
JIN Y, XU Y B, LE P M L, et al. Highly reversible sodium ion batteries enabled by stable electrolyte-electrode interphases[J]. ACS Energy Letters, 2020, 5(10): 3212-3220.
FONDARD J, IRISARRI E, COURRÈGES C, et al. SEI composition on hard carbon in Na-ion batteries after long cycling: Influence of salts (NaPF6, NaTFSI) and additives (FEC, DMCF)[J]. Journal of the Electrochemical Society, 2020, 167(7): doi: 10.1149/1945-7111/ab75fd.
YOON S U, KIM H, JIN H J, et al. Effects of fluoroethylene carbonate-induced solid-electrolyte-interface layers on carbon-based anode materials for potassium ion batteries[J]. Applied Surface Science, 2021, 547: doi: 10.1016/j.apsusc.2021.149193.
BAI P X, HAN X P, HE Y W, et al. Solid electrolyte interphase manipulation towards highly stable hard carbon anodes for sodium ion batteries[J]. Energy Storage Materials, 2020, 25: 324-333.
(a) "Adsorption-intercalation" mechanism<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>; (b) XRD at different potentials<sup>[<xref ref-type="bibr" rid="R10">10</xref>]</sup>; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 2<strong>1.3</strong> 其他储钠机理
... [8]; (b) XRD at different potentials[10]; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation[11]Fig. 2<strong>1.3</strong> 其他储钠机理
... [10];(c) 硬碳在钠化过程中的体积变化以及钠吸附和嵌入[11](a) "Adsorption-intercalation" mechanism<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>; (b) XRD at different potentials<sup>[<xref ref-type="bibr" rid="R10">10</xref>]</sup>; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 2<strong>1.3</strong> 其他储钠机理
... [10]; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation[11]Fig. 2<strong>1.3</strong> 其他储钠机理
... [10];(c) 硬碳在钠化过程中的体积变化以及钠吸附和嵌入[11](a) "Adsorption-intercalation" mechanism<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>; (b) XRD at different potentials<sup>[<xref ref-type="bibr" rid="R10">10</xref>]</sup>; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 2<strong>1.3</strong> 其他储钠机理
... [10]; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation[11]Fig. 2<strong>1.3</strong> 其他储钠机理
... [11](a) "Adsorption-intercalation" mechanism<sup>[<xref ref-type="bibr" rid="R8">8</xref>]</sup>; (b) XRD at different potentials<sup>[<xref ref-type="bibr" rid="R10">10</xref>]</sup>; (c) the volumetric change of the hard carbon in sodiation and illustration of Na-absorption and intercalation<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup>Fig. 2<strong>1.3</strong> 其他储钠机理
... [25]Sodium storage capacity of NPPC and PC (a) Rate capability; (b) Charge-discharge curve of NPPC-2 at 50 mA/g; (c) Long-cycle performance of NPPC-2 at 5 A/g<sup>[<xref ref-type="bibr" rid="R25">25</xref>]</sup>Fig. 3
... [35](a), (b) Initial galvanostatic discharge/charge profiles of all the resulting hard-soft carbon composites at 30 mA/g; (c), (d) The rate capability of all the resulting hard-soft carbon composites; (e), (f) The cycling performance of all the resulting hard-soft carbon composites at 150 mA/g<sup>[<xref ref-type="bibr" rid="R35">35</xref>]</sup>Fig. 4
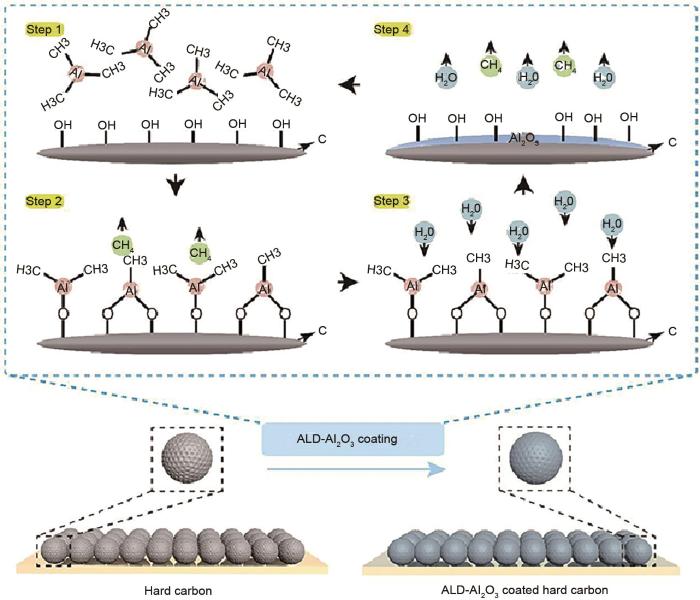
... [37]Schematic of depositing Al<sub>2</sub>O<sub>3</sub> on the surface of the hard carbon electrode<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 5
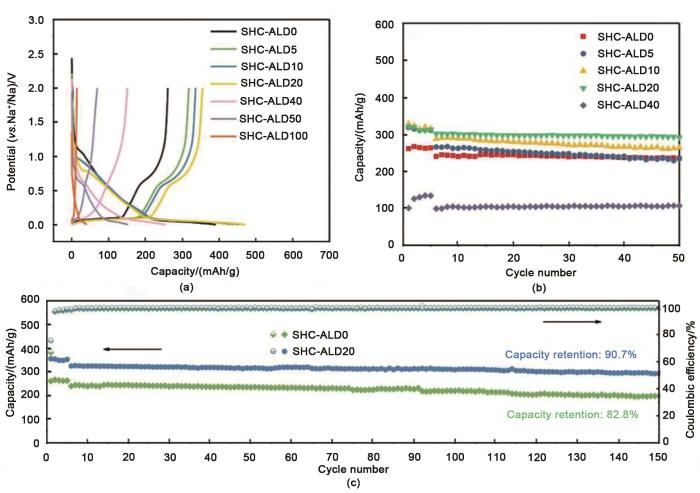
(a) Initial charge-discharge profiles of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at a current density of 20 mA/g; (b) Cycling performance of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at 50 mA/g(The current rate is 20 mA/g in the initial five cycles); (c) Cycling performance of SHC-ALD0 and SHC-ALD20 at a current rate of 50 mA/g(The current rate is 20 mA/g in the initial five cycles)<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 6
(a) Initial charge-discharge profiles of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at a current density of 20 mA/g; (b) Cycling performance of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at 50 mA/g(The current rate is 20 mA/g in the initial five cycles); (c) Cycling performance of SHC-ALD0 and SHC-ALD20 at a current rate of 50 mA/g(The current rate is 20 mA/g in the initial five cycles)<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 6
... [37](a) Initial charge-discharge profiles of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at a current density of 20 mA/g; (b) Cycling performance of hard carbon electrodes coated with different cycles of Al<sub>2</sub>O<sub>3</sub> at 50 mA/g(The current rate is 20 mA/g in the initial five cycles); (c) Cycling performance of SHC-ALD0 and SHC-ALD20 at a current rate of 50 mA/g(The current rate is 20 mA/g in the initial five cycles)<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 6
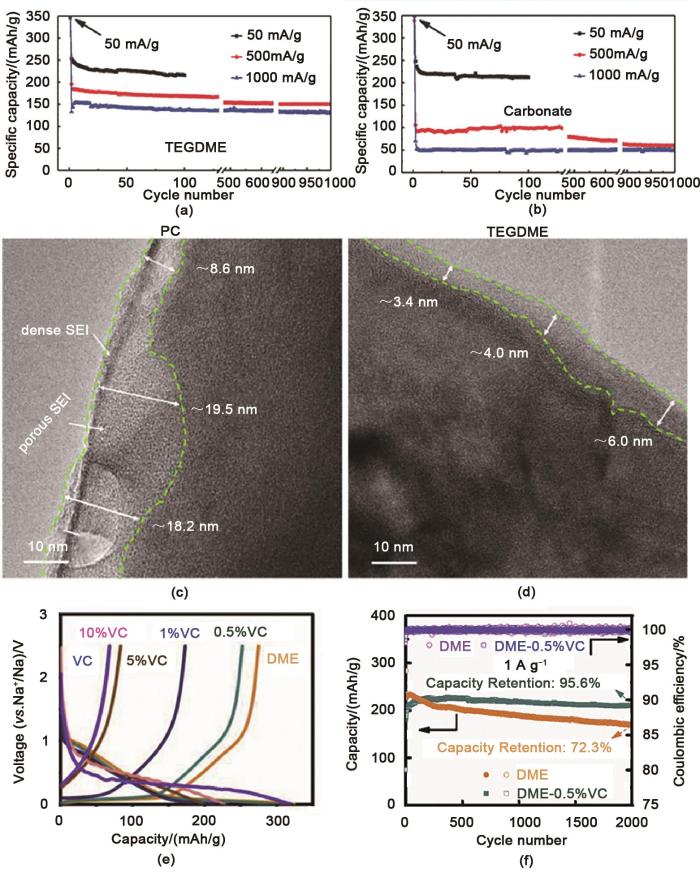
... [40];(b) 碳酸盐中硬碳在不同电流密度下循环性能[40];(c) 通过冷冻透射电镜成像PC电解质中以C/3的速率循环1次后SEI膜厚度[41];(d) 通过冷冻透射电镜成像TEGDME电解质中以C/3的速率循环1次后SEI膜厚度[41];(e) 不同电解质中硬碳充放电曲线[51];(f) 基于DME和DME-0.5%VC电解质的硬碳在电流速率为1A/g下的循环性能[51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论
... [40];(c) 通过冷冻透射电镜成像PC电解质中以C/3的速率循环1次后SEI膜厚度[41];(d) 通过冷冻透射电镜成像TEGDME电解质中以C/3的速率循环1次后SEI膜厚度[41];(e) 不同电解质中硬碳充放电曲线[51];(f) 基于DME和DME-0.5%VC电解质的硬碳在电流速率为1A/g下的循环性能[51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论
... [40]; (b) Cycling performance of HC at different current densities in carbonate[40]; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging[41]; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging[41]; (e) Charge-discharge curves of hard carbon in different electrolytes[51]; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g[51]Fig. 73 结论
... [40]; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging[41]; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging[41]; (e) Charge-discharge curves of hard carbon in different electrolytes[51]; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g[51]Fig. 73 结论
... [41];(d) 通过冷冻透射电镜成像TEGDME电解质中以C/3的速率循环1次后SEI膜厚度[41];(e) 不同电解质中硬碳充放电曲线[51];(f) 基于DME和DME-0.5%VC电解质的硬碳在电流速率为1A/g下的循环性能[51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论
... [41];(e) 不同电解质中硬碳充放电曲线[51];(f) 基于DME和DME-0.5%VC电解质的硬碳在电流速率为1A/g下的循环性能[51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论
... [41]; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging[41]; (e) Charge-discharge curves of hard carbon in different electrolytes[51]; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g[51]Fig. 73 结论
... [41]; (e) Charge-discharge curves of hard carbon in different electrolytes[51]; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g[51]Fig. 73 结论
... [51];(f) 基于DME和DME-0.5%VC电解质的硬碳在电流速率为1A/g下的循环性能[51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论
... [51](a) Cycling performance of HC at different current densities in TEGDME<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (b) Cycling performance of HC at different current densities in carbonate<sup>[<xref ref-type="bibr" rid="R40">40</xref>]</sup>; (c) SEI film thickness after 1 cycle at C/3 rate in PC electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (d) SEI film thickness after 1 cycle at C/3 rate in TEGDME electrolyte by cryo-TEM imaging<sup>[<xref ref-type="bibr" rid="R41">41</xref>]</sup>; (e) Charge-discharge curves of hard carbon in different electrolytes<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>; (f) Cycling performance of hard carbon based on DME and DME-0.5%VC electrolyte at a current rate of 1A/g<sup>[<xref ref-type="bibr" rid="R51">51</xref>]</sup>Fig. 73 结论




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}