[1]
TZIMAS E, FILIOU C, PETEVES S D, et al. Hydrogen storage: State of the art and future perspective[J]. Environmental Science, 2003: 1-91.
[本文引用: 1]
[2]
WIJAYANTA A T, ODA T, PURNOMO C W, et al. Liquid hydrogen, methylcyclohexane, and ammonia as potential hydrogen storage: Comparison review[J]. International Journal of Hydrogen Energy, 2019, 44(29): 15026-15044.
[本文引用: 2]
[3]
ZU L, XU H, ZHANG Q, et al. Design of filament-wound spherical pressure vessels based on non-geodesic trajectories[J]. Composite Structures, 2019, 218: 71-78.
[4]
THOMAS K M. Adsorption and desorption of hydrogen on meta-organic framework materials for storage applications: Comparison with other nanoporous materials[J]. Dalton Transactions, 2009(9): 1487.
[5]
ZHENG J Y, LIU X X, XU P, et al. Development of high pressure gaseous hydrogen storage technologies[J]. International Journal of Hydrogen Energy, 2012, 37(1): 1048-1057.
[6]
WEN J X, MADHAV RAO V C, TAM V H Y. Numerical study of hydrogen explosions in a refuelling environment and in a model storage room[J]. International Journal of Hydrogen Energy, 2010, 35(1): 385-394.
[7]
BINIWALE R B, RAYALU S, DEVOTTA S, et al. Chemical hydrides: A solution to high capacity hydrogen storage and supply[J]. International Journal of Hydrogen Energy, 2008, 33(1): 360-365.
[8]
ZHANG Y, BHATTACHARJEE G, KUMAR R, et al. Solidified hydrogen storage (solid-HyStore) via clathrate hydrates[J]. Chemical Engineering Journal, 2022, 431: doi:10.1016/j.cej.2021.133702.
[本文引用: 2]
[9]
SLOAN E D J, KOH C A, KOH C A. Clathrate hydrates of natural gases[M]. Boca Raton: CRC Press, 2007.
[本文引用: 2]
[10]
ENGLEZOS P. Clathrate hydrates[J]. Industrial & Engineering Chemistry Research, 1993, 32(7): 1251-1274.
[本文引用: 1]
[11]
MORI Y H. Recent advances in hydrate-based technologies for natural gas storage—a review[J]. Journal Fo Chemical Industry and Engineering, 2003, 54(S1): 1-17.
[本文引用: 1]
[12]
DUC N H, CHAUVY F, HERRI J M. CO2 capture by hydrate crystallization-A potential solution for gas emission of steelmaking industry[J]. Energy Conversion and Management, 2007, 48(4): 1313-1322.
[本文引用: 1]
[13]
KANG S P, LEE H E. Recovery of CO2 from flue gas using gas hydrate: Thermodynamic verification through phase equilibrium measurements[J]. Environmental Science & Technology, 2000, 34(20): 4397-4400.
[14]
LINGA P, KUMAR R, ENGLEZOS P. The clathrate hydrate process for post and pre-combustion capture of carbon dioxide[J]. Journal of Hazardous Materials, 2007, 149(3): 625-629.
[15]
LEE S Y, LIANG L Y, RIESTENBERG D, et al. CO2 hydrate composite for ocean carbon sequestration[J]. Environmental Science & Technology, 2003, 37(16): 3701-3708.
[本文引用: 1]
[16]
PARK K N, HONG S Y, LEE J W, et al. A new apparatus for seawater desalination by gas hydrate process and removal characteristics of dissolved minerals (Na+ , Mg2+ , Ca2+ , K+ , B3+ )[J]. Desalination, 2011, 274(1/2/3): 91-96.
[本文引用: 1]
[17]
CHA I, LEE S, LEE J D, et al. Separation of SF6 from gas mixtures using gas hydrate formation[J]. Environmental Science & Technology, 2010, 44(16): 6117-6122.
[本文引用: 1]
[18]
SEO Y, TAJIMA H, YAMASAKI A, et al. A new method for separating HFC-134a from gas mixtures using clathrate hydrate formation[J]. Environmental Science & Technology, 2004, 38(17): 4635-4639.
[本文引用: 1]
[19]
SCHÜTH F. Hydrogen and hydrates[J]. Nature, 2005, 434(7034): 712-713.
[本文引用: 1]
[20]
HU Y H, RUCKENSTEIN E. Clathrate hydrogen hydrate—a promising material for hydrogen storage[J]. Angewandte Chemie (International Ed in English), 2006, 45(13): 2011-2013.
[21]
PATCHKOVSKII S, TSE J S. Thermodynamic stability of hydrogen clathrates[J]. PNAS, 2003, 100(25): 14645-14650.
[本文引用: 1]
[22]
ZHANG Y H, ZHANG P J, YUAN Z M, et al. An investigation on electrochemical hydrogen storage performances of Mg-Y-Ni alloys prepared by mechanical milling[J]. Journal of Rare Earths, 2015, 33(8): 874-883.
[本文引用: 1]
[23]
李璐伶, 樊栓狮, 陈秋雄, 等. 储氢技术研究现状及展望[J]. 储能科学与技术, 2018, 7(4): 586-594.
[本文引用: 1]
LI L L, FAN S S, CHEN Q X, et al. Hydrogen storage technology: Current status and prospects[J]. Energy Storage Science and Technology, 2018, 7(4): 586-594.
[本文引用: 1]
[25]
DYADIN Y A, LARIONOV E G, MANAKOV A Y, et al. Clathrate hydrates of hydrogen and neon[J]. Mendeleev Communications, 1999, 9(5): 209-210.
[本文引用: 1]
[26]
MAO W L. Hydrogen clusters in clathrate hydrate[J]. Science, 2002, 297(5590): 2247-2249.
[本文引用: 1]
[27]
MAO W L, MAO H K. Hydrogen storage in molecular compounds[J]. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(3): 708-710.
[本文引用: 1]
[28]
ENERGY D O. Target explanation document: Onboard hydrogen storage for light-duty fuel cell vehicles[M]. the US DRIVE Partnership, 2017.
[本文引用: 1]
[29]
OGATA K, HASHIMOTO S, SUGAHARA T, et al. Storage capacity of hydrogen in tetrahydrofuran hydrate[J]. Chemical Engineering Science, 2008, 63(23): 5714-5718.
[本文引用: 7]
[30]
DI PROFIO P, CANALE V, GERMANI R, et al. Reverse micelles enhance the formation of clathrate hydrates of hydrogen[J]. Journal of Colloid and Interface Science, 2018, 516: 224-231.
[本文引用: 2]
[31]
TSUDA T, OGATA K, HASHIMOTO S, et al. Storage capacity of hydrogen in tetrahydrothiophene and furan clathrate hydrates[J]. Chemical Engineering Science, 2009, 64(19): 4150-4154.
[本文引用: 5]
[32]
TRUEBA A T, RADOVIĆ I R, ZEVENBERGEN J F, et al. Kinetic measurements and in situ Raman spectroscopy study of the formation of TBAF semi-hydrates with hydrogen and carbon dioxide[J]. International Journal of Hydrogen Energy, 2013, 38(18): 7326-7334.
[本文引用: 3]
[33]
ANDERSON R, CHAPOY A, TOHIDI B. Phase relations and binary clathrate hydrate formation in the system H2 -THF-H2 O[J]. Langmuir: the ACS Journal of Surfaces and Colloids, 2007, 23(6): 3440-3444.
[本文引用: 4]
[34]
TRUEBA A T, ROVETTO L J, FLORUSSE L J, et al. Phase equilibrium measurements of structure II clathrate hydrates of hydrogen with various promoters[J]. Fluid Phase Equilibria, 2011, 307(1): 6-10.
[35]
SAKAMOTO J, HASHIMOTO S, TSUDA T, et al. Thermodynamic and Raman spectroscopic studies on hydrogen+tetra-n-butyl ammonium fluoride semi-clathrate hydrates[J]. Chemical Engineering Science, 2008, 63(24): 5789-5794.
[本文引用: 4]
[36]
KASHCHIEV D, FIROOZABADI A. Induction time in crystallization of gas hydrates[J]. Journal of Crystal Growth, 2003, 250(3/4): 499-515.
[本文引用: 1]
[37]
SLOAN E D, FLEYFEL F. A molecular mechanism for gas hydrate nucleation from ice[J]. AIChE Journal, 1991, 37(9): 1281-1292.
[本文引用: 1]
[38]
RADHAKRISHNAN R, TROUT B L. A new approach for studying nucleation phenomena using molecular simulations: Application to CO2 hydrate clathrates[J]. The Journal of Chemical Physics, 2002, 117(4): 1786-1796.
[本文引用: 1]
[39]
JACOBSON L C, HUJO W, MOLINERO V. Amorphous precursors in the nucleation of clathrate hydrates[J]. Journal of the American Chemical Society, 2010, 132(33): 11806-11811.
[本文引用: 1]
[40]
陈光进, 孙长宇, 马庆兰. 气体水合物科学与技术[M]. 北京: 化学工业出版社, 2008.
[本文引用: 1]
CHEN G J, SUN C Y, MA Q L. Gas hydrate science and technology [M]. Beijing: Chemical Industry Press, 2008.
[本文引用: 1]
[41]
DU J, WANG L, LIANG D, et al. Phase equilibria and dissociation enthalpies of hydrogen semi-clathrate hydrate with tetrabutyl ammonium nitrate[J]. Journal of Chemical & Engineering Data, 2012, 57(2): 603-609.
[本文引用: 2]
[42]
LUNINE J I, STEVENSON D J. Thermodynamics of clathrate hydrate at low and high pressures with application to the outer solar system[J]. The Astrophysical Journal Letters Supplement Series, 1985, 58: 493.
[本文引用: 1]
[43]
ZHONG J R, CHEN L T, LIU T C, et al. Sieving of hydrogen-containing gas mixtures with tetrahydrofuran hydrate[J]. The Journal of Physical Chemistry C, 2017, 121(50): 27822-27829.
[本文引用: 1]
[44]
VLASOV V A. Diffusion model of gas hydrate formation from ice[J]. Heat and Mass Transfer, 2016, 52(3): 531-537.
[本文引用: 1]
[45]
DARTOIS E, LANGLET F. Carbon dioxide clathrate hydrate formation at low temperature[J]. Astronomy & Astrophysics, 2021, 652: A74.
[本文引用: 1]
[46]
TRUEBA A T, RADOVIĆ I R, ZEVENBERGEN J F, et al. Kinetics measurements and in situ Raman spectroscopy of formation of hydrogen-tetrabutylammonium bromide semi-hydrates[J]. International Journal of Hydrogen Energy, 2012, 37(7): 5790-5797.
[本文引用: 2]
[47]
VELUSWAMY H P, LINGA P. Macroscopic kinetics of hydrate formation of mixed hydrates of hydrogen/tetrahydrofuran for hydrogen storage[J]. International Journal of Hydrogen Energy, 2013, 38(11): 4587-4596.
[本文引用: 4]
[48]
SUGAHARA T, HAAG J C, PRASAD P S R, et al. Increasing hydrogen storage capacity using tetrahydrofuran[J]. Journal of the American Chemical Society, 2009, 131(41): 14616-14617.
[本文引用: 2]
[49]
SUGAHARA T, HAAG J C, WARNTJES A A, et al. Large-cage occupancies of hydrogen in binary clathrate hydrates dependent on pressures and guest concentrations[J]. The Journal of Physical Chemistry C, 2010, 114(35): 15218-15222.
[本文引用: 3]
[50]
SHIN K, KIM Y, STROBEL T A, et al. Tetra-n-butylammonium borohydride semiclathrate: A hybrid material for hydrogen storage[J]. The Journal of Physical Chemistry A, 2009, 113(23): 6415-6418.
[本文引用: 1]
[51]
KUMAR R, KLUG D D, RATCLIFFE C I, et al. Low-pressure synthesis and characterization of hydrogen-filled ice Ic[J]. Angewandte Chemie (International Ed in English), 2013, 52(5): 1531-1534.
[本文引用: 1]
[52]
STRÖBEL R, GARCHE J, MOSELEY P T, et al. Hydrogen storage by carbon materials[J]. Journal of Power Sources, 2006, 159(2): 781-801.
[本文引用: 1]
[53]
STROBEL T A, KOH C A, SLOAN E D. Hydrogen storage properties of clathrate hydrate materials[J]. Fluid Phase Equilibria, 2007, 261(1/2): 382-389.
[本文引用: 1]
[54]
YOSHIOKA H, OTA M, SATO Y, et al. Decomposition kinetics and recycle of binary hydrogen-tetrahydrofuran clathrate hydrate[J]. AIChE Journal, 2011, 57(1): 265-272.
[本文引用: 1]
[55]
NAGAI Y, YOSHIOKA H, OTA M, et al. Binary hydrogen-tetrahydrofuran clathrate hydrate formation kinetics and models[J]. AIChE Journal, 2008, 54(11): 3007-3016.
[本文引用: 2]
[56]
邓灿, 梁德青, 李栋梁. 环戊烷-氢气水合物形成过程研究[J]. 石油化工, 2009, 38(9): 951-956.
[本文引用: 2]
DENG C, LIANG D Q, LI D L. Formation of cyclopentane-hydrogen clathrate hydrates[J]. Petrochemical Technology, 2009, 38(9): 951-956.
[本文引用: 2]
[57]
SU F B, BRAY C L, CARTER B O, et al. Reversible hydrogen storage in hydrogel clathrate hydrates[J]. Advanced Materials, 2009, 21(23): 2382-2386.
[本文引用: 1]
[58]
GHAANI M R, SCHICKS J M, ENGLISH N J. A review of reactor designs for hydrogen storage in clathrate hydrates[J]. Applied Sciences, 2021, 11(2): 469.
[本文引用: 1]
[59]
IWAI Y, HIRATA M. Molecular dynamics simulation of diffusion of hydrogen in binary hydrogen-tetrahydrofuran hydrate[J]. Molecular Simulation, 2012, 38(4): 333-340.
[本文引用: 1]
[60]
HASEGAWA T, BRUMBY P E, YASUOKA K, et al. Mechanism for H2 diffusion in sII hydrates by molecular dynamics simulations[J]. The Journal of Chemical Physics, 2020, 153(5): 054706.
[本文引用: 1]
[61]
GORMAN P D, ENGLISH N J, MACELROY J M D. Dynamical cage behaviour and hydrogen migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates[J]. The Journal of Chemical Physics, 2012, 136(4): 044506.
[本文引用: 1]
[62]
WANG Y H, YIN K D, LANG X M, et al. Hydrogen storage in sH binary hydrate: Insights from molecular dynamics simulation[J]. International Journal of Hydrogen Energy, 2021, 46(29): 15748-15760.
[本文引用: 2]
[63]
WANG Y H, YIN K D, FAN S S, et al. The molecular insight into the "Zeolite-ice" as hydrogen storage material[J]. Energy, 2021, 217: doi:10.1016/j.energy.2020.119406.
[本文引用: 2]
[64]
DU J W, LIANG D Q, LI D L, et al. Experimental determination of the equilibrium conditions of binary gas hydrates of cyclopentane + oxygen, cyclopentane + nitrogen, and cyclopentane + hydrogen[J]. Industrial & Engineering Chemistry Research, 2010, 49(22): 11797-11800.
[本文引用: 3]
[65]
DUARTE A R C, SHARIATI A, PETERS C J. Phase equilibrium measurements of structure sH hydrogen clathrate hydrates with various promoters[J]. Journal of Chemical & Engineering Data, 2009, 54(5): 1628-1632.
[本文引用: 1]
[66]
LIU J X, YAN Y J, CHEN G, et al. Prediction of efficient promoter molecules of sH hydrogen hydrate: An ab initio study[J]. Chemical Physics, 2019, 516: 15-21.
[本文引用: 1]
[67]
KOMATSU H, YOSHIOKA H, OTA M, et al. Phase equilibrium measurements of hydrogen–tetrahydrofuran and hydrogen–cyclopentane binary clathrate hydrate systems[J]. Journal of Chemical & Engineering Data, 2010, 55(6): 2214-2218.
[本文引用: 3]
[68]
BURNHAM C J, FUTERA Z, ENGLISH N J. Quantum and classical inter-cage hopping of hydrogen molecules in clathrate hydrate: Temperature and cage-occupation effects[J]. Physical Chemistry Chemical Physics: PCCP, 2016, 19(1): 717-728.
[本文引用: 1]
[69]
邓灿. 氢气水合物形成过程研究 [D]. 广州: 中国科学院广州能源研究所, 2009.
[本文引用: 1]
Deng C. Study on the hydrogen hydrate formation[D]. Guangzhou: Guangzhou Institute of Energy, Chinese Academy of Science. 2009.
[本文引用: 1]
[70]
TRINH T T, WAAGE M H, VAN ERP T S, et al. Low barriers for hydrogen diffusion in sII clathrate[J]. Physical Chemistry Chemical Physics: PCCP, 2015, 17(21): 13808-13812
[本文引用: 1]
[71]
HE Y, SUN M T, CHEN C, et al. Surfactant-based promotion to gas hydrate formation for energy storage[J]. Journal of Materials Chemistry A, 2019, 7(38): 21634-21661.
[本文引用: 1]
[72]
VELUSWAMY H P, ANG W J, ZHAO D, et al. Influence of cationic and non-ionic surfactants on the kinetics of mixed hydrogen/tetrahydrofuran hydrates[J]. Chemical Engineering Science, 2015, 132: 186-199.
[本文引用: 1]
[73]
VELUSWAMY H P, CHEN J Y, LINGA P. Surfactant effect on the kinetics of mixed hydrogen/propane hydrate formation for hydrogen storage as clathrates[J]. Chemical Engineering Science, 2015, 126: 488-499.
[本文引用: 1]
[74]
ZHANG Y, BHATTACHARJEE G, ZHENG J J, et al. Hydrogen storage as clathrate hydrates in the presence of 1, 3-dioxolane as a dual-function promoter[J]. Chemical Engineering Journal, 2022, 427: doi: 10.1016/j.cej.2021.131771.
[本文引用: 1]
[75]
YU C, FAN S S, LANG X M, et al. Hydrogen and chemical energy storage in gas hydrate at mild conditions[J]. International Journal of Hydrogen Energy, 2020, 45(29): 14915-14921.
[本文引用: 1]
[76]
FANG Y J, XIE Y M, ZHOU X F, et al. Influence of activated carbon to the hydrogen storage characteristics of THF hydrate[J]. Advanced Materials Research, 2014, 887/888: 493-496.
[本文引用: 1]
[77]
吕秋楠, 宋永臣, 李小森. 鼓泡器中环戊烷-甲烷-盐水体系水合物的生成动力学[J]. 化工进展, 2016, 35(12): 3777-3782.
[本文引用: 1]
LÜ Q N, SONG Y C, LI X S. Formation kinetics of cyclopentane-methane hydrate in NaCl solution with a bubbling equipment[J]. Chemical Industry and Engineering Progress, 2016, 35(12): 3777-3782.
[本文引用: 1]
[78]
LI G, LIU D P, XIE Y M, et al. Study on effect factors for CO2 hydrate rapid formation in a water-spraying apparatus[J]. Energy & Fuels, 2010, 24(8): 4590-4597.
[本文引用: 1]
[79]
SAHA D, DENG S G. Accelerated formation of THF-H2 clathrate hydrate in porous media[J]. Langmuir: the ACS Journal of Surfaces and Colloids, 2010, 26(11): 8414-8418.
[本文引用: 1]
[80]
SU F B, BRAY C L, TAN B E, et al. Rapid and reversible hydrogen storage in clathrate hydrates using emulsion-templated polymers[J]. Advanced Materials, 2008, 20(14): 2663-2666.
[本文引用: 1]
[81]
ZHAO W H, WANG L, BAI J, et al. Spontaneous formation of one-dimensional hydrogen gas hydrate in carbon nanotubes[J]. Journal of the American Chemical Society, 2014, 136(30): 10661-10668.
[本文引用: 1]
[82]
LEE H E, LEE J W, KIM D Y, et al. Tuning clathrate hydrates for hydrogen storage[J]. Nature, 2005, 434(7034): 743-746.
[本文引用: 2]
[83]
KIM D Y, PARK J, LEE J W, et al. Critical guest concentration and complete tuning pattern appearing in the binary clathrate hydrates[J]. Journal of the American Chemical Society, 2006, 128(48): 15360-15361.
[本文引用: 1]
[84]
ROMáN-PéREZ G, MOAIED M, SOLER J M,et al. Stability, adsorption,and diffusion of CH4 , CO2 , and H2 in clathrate hydrates[J]. Physical Review Letters,2010, 105: doi: 10.1103/PhysRevLett.105.145901.
[本文引用: 1]
1
... 在资源环境问题突出和“碳达峰、碳中和”的时代背景下,寻求发展高效、清洁可持续的能源成为我国能源发展领域的重要议题.氢能是典型的绿色能源,具有燃烧热值高的特点,是化石能源3~5倍[1 ] ,成为了可替代三大化石能源的经济、有效的新能源之一. ...
2
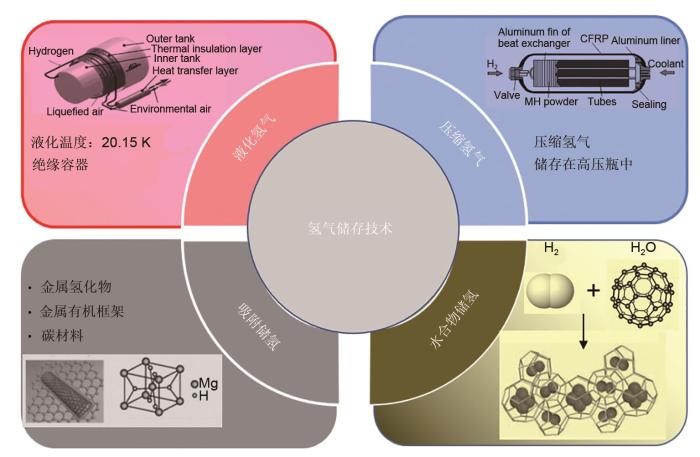
... 氢能储存技术作为氢能从生产到利用的中间桥梁,是氢能发展不可或缺的一部分.氢气储存主要分为物理储存和化学储存,如图1 所示,常见的方法有低温液化储氢、高压气态储氢、吸附储氢、金属合金储氢、水合物储氢等[2 -8 ] .低温液化需要在常压下将氢气降温到20 K,液化1 kg氢气需要消耗15.2 kWh的电能[2 ] ,相当于储存氢气能量的30%.同时储运过程中对于容器的绝热性能要求极高,成本较高.高压储氢则是在常温下将气态的氢压缩至高压状态储存在气罐中,应用比较广泛,成本低.吸附储氢是利用材料与氢气分子间的相互作用将氢气吸附到固体物质载体中,这在一定程度上限制了储存容量;金属合金储氢条件较为温和,但需要高温进行释氢,因此需要额外的能量供给. ...
... [2 ],相当于储存氢气能量的30%.同时储运过程中对于容器的绝热性能要求极高,成本较高.高压储氢则是在常温下将气态的氢压缩至高压状态储存在气罐中,应用比较广泛,成本低.吸附储氢是利用材料与氢气分子间的相互作用将氢气吸附到固体物质载体中,这在一定程度上限制了储存容量;金属合金储氢条件较为温和,但需要高温进行释氢,因此需要额外的能量供给. ...
2
... 氢能储存技术作为氢能从生产到利用的中间桥梁,是氢能发展不可或缺的一部分.氢气储存主要分为物理储存和化学储存,如图1 所示,常见的方法有低温液化储氢、高压气态储氢、吸附储氢、金属合金储氢、水合物储氢等[2 -8 ] .低温液化需要在常压下将氢气降温到20 K,液化1 kg氢气需要消耗15.2 kWh的电能[2 ] ,相当于储存氢气能量的30%.同时储运过程中对于容器的绝热性能要求极高,成本较高.高压储氢则是在常温下将气态的氢压缩至高压状态储存在气罐中,应用比较广泛,成本低.吸附储氢是利用材料与氢气分子间的相互作用将氢气吸附到固体物质载体中,这在一定程度上限制了储存容量;金属合金储氢条件较为温和,但需要高温进行释氢,因此需要额外的能量供给. ...
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...
2
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
... 添加剂的使用使水合物储氢具有了热力学可行性,然而,气体水合物的生成过程是一个多相复杂体系内气体分子和水分子相互作用的动力学过程,是其流体相向固体相转变的过程,更像是一个结晶动力学过程,包括成核和生长两个动力学过程[9 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 笼型水合物是一种在特定温度和压力条件下形成的非化学计量晶体化合物,主体水分子通过氢键形成笼型结构,客体分子与水分子之间的范德华力使客体分子填充到空笼中形成稳定的物质.常见的客体分子有烷烃类气体、二氧化碳、四氢呋喃、氢气等[9 -10 ] .基于笼型水合物具有独特的结构和理化性质,应用越来越广泛,主要有天然气储运[11 ] 、二氧化碳捕获[12 -15 ] 、海水淡化[16 ] 、气体分离[17 -18 ] 、储氢[19 -21 ] 等技术.水合物储氢作为一种固态储氢技术,发展相对较晚,但因其储氢释氢过程完全可逆、分子形式储存、相对高的单位储氢量、安全性高、不易爆等特点被视为21世纪氢能储存最具发展潜力的技术[22 -23 ] . ...
1
... 早期,研究人员认为氢气分子尺寸太小,不能形成水合物.直到1993年,Vos等[24] 首次发现在压力0.75 GPa和3.1 GPa,温度为295 K的情况下可以形成氢气水合物,推翻了氢气分子不能稳定存在于水合物笼中的猜测.随后Dyadin[25 ] 、Mao等[26 ] 分别通过一系列实验和技术证实了氢气水合物的结构,并基于此提出了水合物储氢的概念;随后Mao等[27 ] 在240~249 K、200~300 MPa条件下得到了5.3%(质量分数,下同)的储氢量的氢气水合物,满足DOE对于车载氢燃料储氢密度的要求[28 ] ,从此水合物才作为储氢材料进入人们的视野. ...
1
... 早期,研究人员认为氢气分子尺寸太小,不能形成水合物.直到1993年,Vos等[24] 首次发现在压力0.75 GPa和3.1 GPa,温度为295 K的情况下可以形成氢气水合物,推翻了氢气分子不能稳定存在于水合物笼中的猜测.随后Dyadin[25 ] 、Mao等[26 ] 分别通过一系列实验和技术证实了氢气水合物的结构,并基于此提出了水合物储氢的概念;随后Mao等[27 ] 在240~249 K、200~300 MPa条件下得到了5.3%(质量分数,下同)的储氢量的氢气水合物,满足DOE对于车载氢燃料储氢密度的要求[28 ] ,从此水合物才作为储氢材料进入人们的视野. ...
1
... 早期,研究人员认为氢气分子尺寸太小,不能形成水合物.直到1993年,Vos等[24] 首次发现在压力0.75 GPa和3.1 GPa,温度为295 K的情况下可以形成氢气水合物,推翻了氢气分子不能稳定存在于水合物笼中的猜测.随后Dyadin[25 ] 、Mao等[26 ] 分别通过一系列实验和技术证实了氢气水合物的结构,并基于此提出了水合物储氢的概念;随后Mao等[27 ] 在240~249 K、200~300 MPa条件下得到了5.3%(质量分数,下同)的储氢量的氢气水合物,满足DOE对于车载氢燃料储氢密度的要求[28 ] ,从此水合物才作为储氢材料进入人们的视野. ...
1
... 早期,研究人员认为氢气分子尺寸太小,不能形成水合物.直到1993年,Vos等[24] 首次发现在压力0.75 GPa和3.1 GPa,温度为295 K的情况下可以形成氢气水合物,推翻了氢气分子不能稳定存在于水合物笼中的猜测.随后Dyadin[25 ] 、Mao等[26 ] 分别通过一系列实验和技术证实了氢气水合物的结构,并基于此提出了水合物储氢的概念;随后Mao等[27 ] 在240~249 K、200~300 MPa条件下得到了5.3%(质量分数,下同)的储氢量的氢气水合物,满足DOE对于车载氢燃料储氢密度的要求[28 ] ,从此水合物才作为储氢材料进入人们的视野. ...
7
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
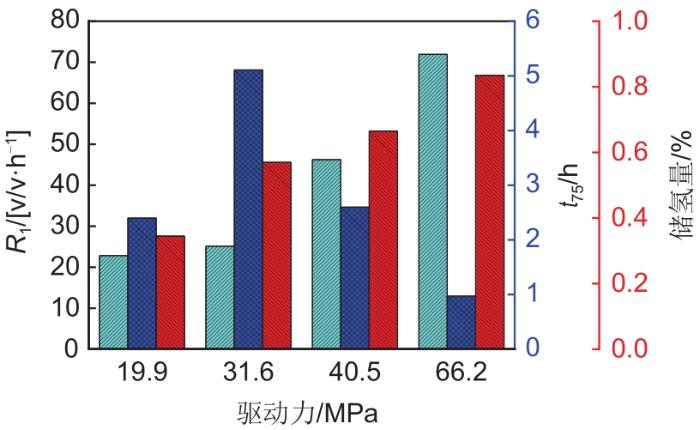
... 驱动力是影响氢气分子扩散的关键因素,图3 是在277.15 K不同驱动力下水合储氢实验数据图.如图3 所示,提高驱动力,水合储氢的初始反应速率(R 1 )大幅提升,从22.84 v/v· h-1 提升到71.93 v/v· h-1 ,生成时间随之减少,同时储氢密度提高.当驱动力从19.9 MPa增加到66.2 MPa时,储氢量从0.345%增加到0.835%[29 ] ,小笼占据率达到了79%.可以看出驱动力是强化氢气分子扩散,促进氢气水合物成核和生长的关键因素. ...
... [
29 ]
Graph showing hydrogen hydrate storage with different driver forces under 277.15 K<sup>[<xref ref-type="bibr" rid="R28">29</xref>]</sup> Fig. 3 ![]()
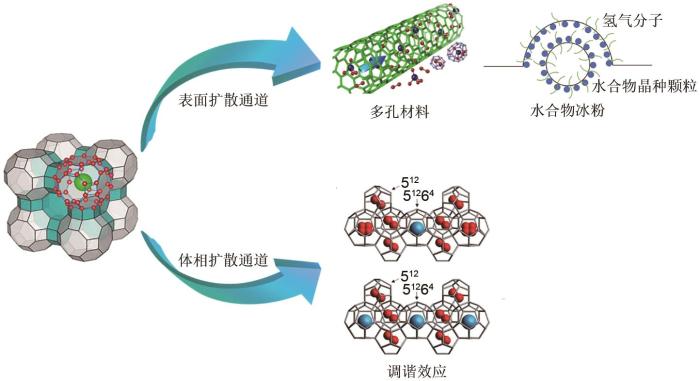
此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
... [
29 ]
Fig. 3 ![]()
此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
... 除使用热力学促进剂之外,改变温度压力也是提高水合储氢性能的有效方法.表5 为不同压力下水合储氢动力学参数汇总.Trueba等[31 ] 以5.0%(摩尔分数)四氢噻吩(THT)和呋喃为促进剂,研究了二元氢气水合物与压力的关系,由表5 可知,当温度稳定在277.15 K时,压力驱动力△P 从12.0 MPa增加到28.7 MPa时,水合物生长阶段前1小时的气体吸收速率大幅提升,初始动力学速率提高了93.8%~170.0%;而t 75 随压力变化较少,储氢密度相对提高,证明提高压力可以在初始阶段维持了相对较高的生长速率,压力是促进氢气水合物成核和生长的关键因素.Ogata等[29 ] 研究了5.6%(摩尔分数)THF在11.4~66.4 MPa下水合物的储氢性能,结果表明随着压力的提高,储氢速率和储氢量明显提高,其中压力主要影响反应初期前1 h内的储氢速率,后期总体反应时间相差不大,并且随着压力的增加,储氢密度增长趋势减慢,呈现饱和状态.Sugahara等[49 ] 通过拉曼光谱、X射线衍射分析以及分解测试研究了氢气分子在THF水合物中的占据情况,结果发现即使压力提升至74 MPa,水合物储氢密度也不会显著增长,始终维持在0.7%~1%这说明提高压力增大驱动力可以提高水合储氢性能,但达到一定压力后会出现边界效应,即水合储氢性能不会出现明显改善;Burnham等[68 ] 的分子动力学模拟认为sII氢气水合物的大笼占据率很少会超过两个,在100 MPa下小笼512 的平均占据为0.85~1更符合现实,这也解释了边界效应的原因. ...
... 在275.15 K 下不同压力驱动力下水合储氢实验数据汇总表[29 , 31 , 49 ] ...
2
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
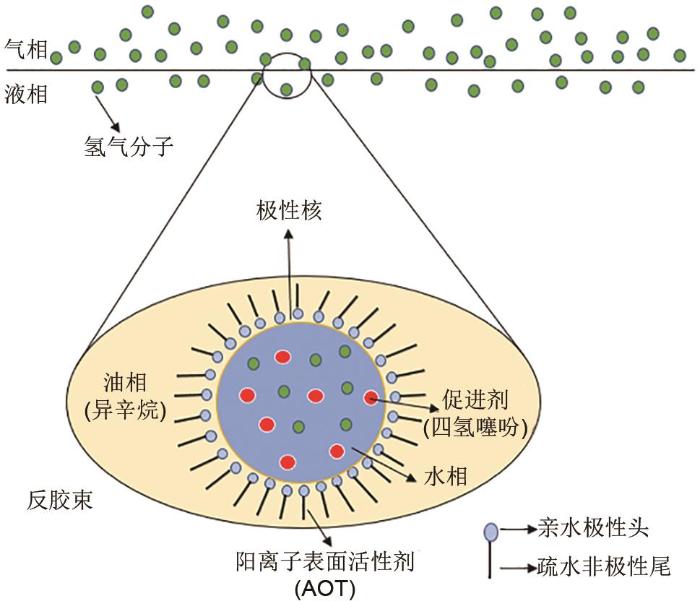
... 表面活性剂可以在气液两相界面吸附形成胶束降低水的表面张力,也可以吸附在液体界面间形成反胶束来降低油水界面张力.Di等[30 ] 以200 mL异辛烷为分散介质,溶解适量2-乙基己基琥珀酸酯磺酸钠(AOT)形成0.1 mol/L的反胶束体系,如图6 所示.在以四氢噻吩为促进剂的体系下,与原本数十个小时的生成过程相比,水合物生成动力学速率明显提升,诱导时间为3 min,水合物生成时间仅为28 min,最大储氢量为0.5%.反胶束是将表面活性剂溶于非极性的有机溶剂中,当浓度超过临界胶束浓度时在有机溶剂内形成的胶束;反胶束作用机理在于表面活性剂的非极性尾向外伸入非极性有机溶剂主体中,而极性头则向内排列形成一个极性核,此极性核具有溶解促进剂分子的能力,可以有效应用到添加了热力学添加剂的氢气水合物体系.并且该研究还发现反胶束可以增强水与非水溶性分子的接触,稳定形成包裹促进剂的纳米水滴,从而增大气液接触面积,因此反胶束对于环戊烷(CP)、四氢噻吩(THT)等非水溶性促进剂的储氢效果更加明显. ...
5
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 除使用热力学促进剂之外,改变温度压力也是提高水合储氢性能的有效方法.表5 为不同压力下水合储氢动力学参数汇总.Trueba等[31 ] 以5.0%(摩尔分数)四氢噻吩(THT)和呋喃为促进剂,研究了二元氢气水合物与压力的关系,由表5 可知,当温度稳定在277.15 K时,压力驱动力△P 从12.0 MPa增加到28.7 MPa时,水合物生长阶段前1小时的气体吸收速率大幅提升,初始动力学速率提高了93.8%~170.0%;而t 75 随压力变化较少,储氢密度相对提高,证明提高压力可以在初始阶段维持了相对较高的生长速率,压力是促进氢气水合物成核和生长的关键因素.Ogata等[29 ] 研究了5.6%(摩尔分数)THF在11.4~66.4 MPa下水合物的储氢性能,结果表明随着压力的提高,储氢速率和储氢量明显提高,其中压力主要影响反应初期前1 h内的储氢速率,后期总体反应时间相差不大,并且随着压力的增加,储氢密度增长趋势减慢,呈现饱和状态.Sugahara等[49 ] 通过拉曼光谱、X射线衍射分析以及分解测试研究了氢气分子在THF水合物中的占据情况,结果发现即使压力提升至74 MPa,水合物储氢密度也不会显著增长,始终维持在0.7%~1%这说明提高压力增大驱动力可以提高水合储氢性能,但达到一定压力后会出现边界效应,即水合储氢性能不会出现明显改善;Burnham等[68 ] 的分子动力学模拟认为sII氢气水合物的大笼占据率很少会超过两个,在100 MPa下小笼512 的平均占据为0.85~1更符合现实,这也解释了边界效应的原因. ...
... 在275.15 K 下不同压力驱动力下水合储氢实验数据汇总表[29 , 31 , 49 ] ...
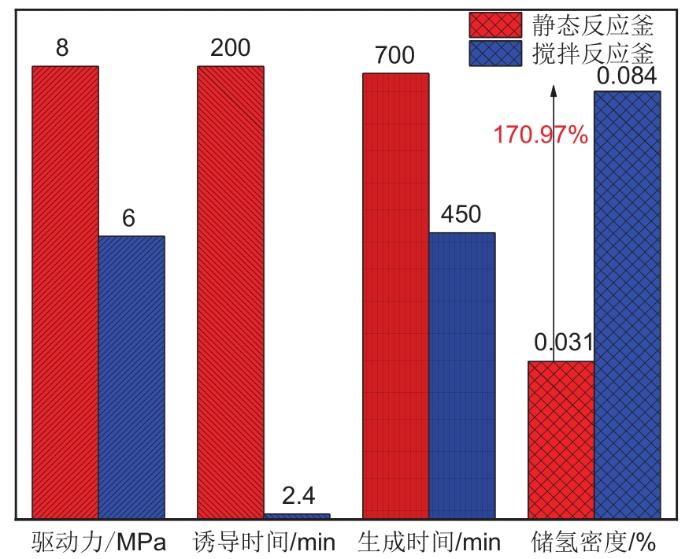
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
3
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
... 对比近年来水合储氢相关实验结果(表3 ),可以发现,当水合物样品量极小时,储氢密度可以达到3%以上,而当样品量超过5 g时,储氢量就只有0.5%左右.随样品量的增加,达到100 g以上时[32 ,46 -47 ] ,储量仅剩0.1%.这些结果表明即使氢气分子进出水合物笼相较甲烷等大分子气体容易,但其储氢速率和储氢密度仍受气液接触面积以及氢气分子在水合物相中扩散的影响. ...
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
4
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
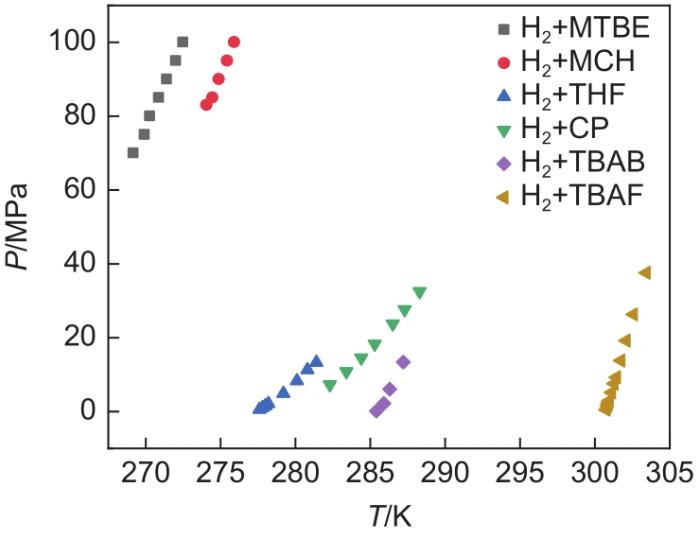
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
... [
33 ,
35 ,
64 -
67 ]
Phase diagram showing different promoters of the binary hydrogen hydrate<sup>[<xref ref-type="bibr" rid="R32">33</xref>, <xref ref-type="bibr" rid="R34">35</xref>, <xref ref-type="bibr" rid="R63">64</xref>-<xref ref-type="bibr" rid="R66">67</xref>]</sup> Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
... [
33 ,
35 ,
64 -
67 ]
Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
4
... 然而氢气水合物生成条件过于苛刻,几乎不具有工业化应用的可能性.为此研究人员通过大量的研究发现向氢气水合物体系内引入某些添加剂可以大幅降低氢气水合物的相平衡条件,这类添加剂被称为热力学添加剂,常见的热力学添加剂有四氢呋喃[29 ] 、环戊烷[30 ] 、呋喃[31 ] 、四丁基氟化铵[32 ] 等.添加剂通过占据水合物笼的空腔稳定水合物结构,从而满足降低相平衡条件的要求.利用添加剂可将氢气水合物的相平衡从200 MPa、270 K降到10 MPa、280.5~301.5 K[33 -35 ] .添加剂的应用使水合物储氢具有工业化前景. ...
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
... ,
35 ,
64 -
67 ]
Phase diagram showing different promoters of the binary hydrogen hydrate<sup>[<xref ref-type="bibr" rid="R32">33</xref>, <xref ref-type="bibr" rid="R34">35</xref>, <xref ref-type="bibr" rid="R63">64</xref>-<xref ref-type="bibr" rid="R66">67</xref>]</sup> Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
... ,
35 ,
64 -
67 ]
Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
1
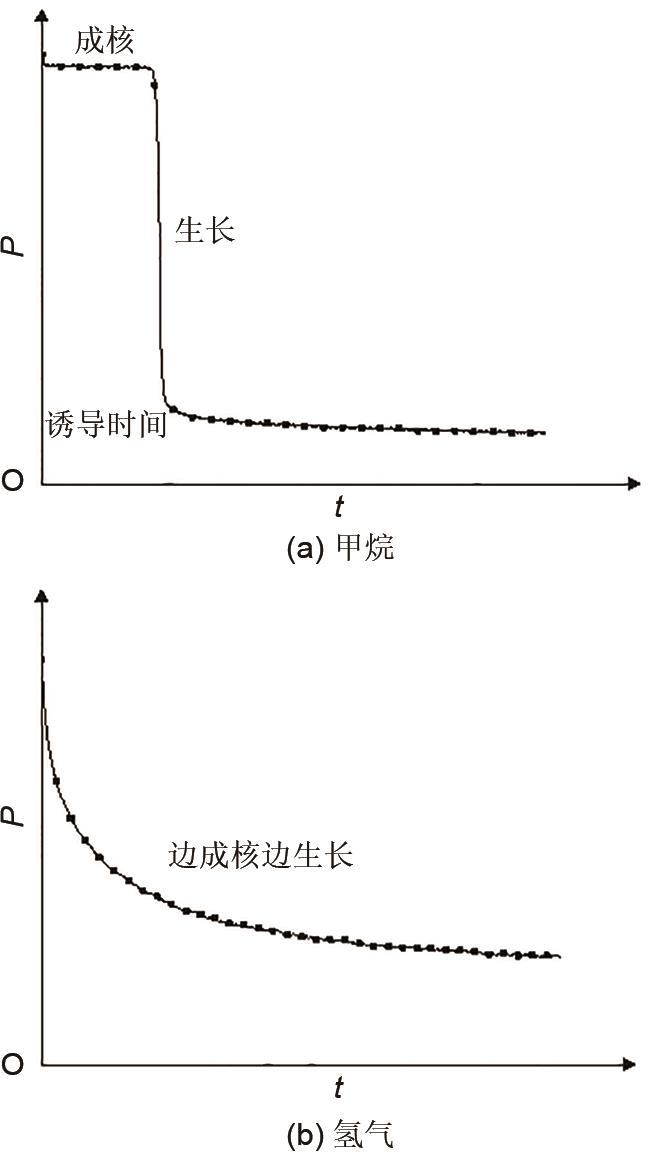
... ② 渐进成核:水合物边成核边生长,生长过程中伴随着成核,即水合物生长过程中晶粒数目是逐步增多的.当发生渐进成核时,成核速率与时间无关,并且符合公式(1) ,渐进成核速率[36 ] 由式(1) 给出. ...
1
... 从相态来说水合物成核也可以分为均质成核和非均质成核.均质成核是指纯液体在没有其他任何杂质情况下的成核过程;非均质成核是指在溶液中存在其他物质的情况,这种成核过程在现实中更加普遍.目前有多种水合物成核模型来解释非均质水合物成核的微观机理,主要有成簇成核模型[37 ] 、界面成核模型[38 ] 、团簇成核模型[39 ] 等,他们普遍认为水合物成核一般发生在气液界面,界面处所需的成核吉布斯自由能较小,并且由于界面处的主体分子浓度高,气液混合引起界面的晶体结构向内部扩散,从而导致大量晶核的出现. ...
1
... 从相态来说水合物成核也可以分为均质成核和非均质成核.均质成核是指纯液体在没有其他任何杂质情况下的成核过程;非均质成核是指在溶液中存在其他物质的情况,这种成核过程在现实中更加普遍.目前有多种水合物成核模型来解释非均质水合物成核的微观机理,主要有成簇成核模型[37 ] 、界面成核模型[38 ] 、团簇成核模型[39 ] 等,他们普遍认为水合物成核一般发生在气液界面,界面处所需的成核吉布斯自由能较小,并且由于界面处的主体分子浓度高,气液混合引起界面的晶体结构向内部扩散,从而导致大量晶核的出现. ...
1
... 从相态来说水合物成核也可以分为均质成核和非均质成核.均质成核是指纯液体在没有其他任何杂质情况下的成核过程;非均质成核是指在溶液中存在其他物质的情况,这种成核过程在现实中更加普遍.目前有多种水合物成核模型来解释非均质水合物成核的微观机理,主要有成簇成核模型[37 ] 、界面成核模型[38 ] 、团簇成核模型[39 ] 等,他们普遍认为水合物成核一般发生在气液界面,界面处所需的成核吉布斯自由能较小,并且由于界面处的主体分子浓度高,气液混合引起界面的晶体结构向内部扩散,从而导致大量晶核的出现. ...
1
... 水合物的生成需要低温环境,而水合物的生成又是个放热反应,大量生成热的释放会抑制水合物的成核以及生长,甚至造成水合物的分解.对于sII型二元氢气水合物,其分解焓大小与相平衡条件、添加剂分子以及水合物中水分子间的氢键强弱等因素相关.表1 给出了不同体系下二元水合物的分解焓计算结果.从表1 可以看出,水合储氢过程中的水合物热不能忽略.氢气水合物的渐进式成核生长虽不会大幅提高体系温度,造成热抑制[40 ] .但若这些热不能及时排除,仍会使体系温度提高从而削弱水合反应速率. ...
1
... 水合物的生成需要低温环境,而水合物的生成又是个放热反应,大量生成热的释放会抑制水合物的成核以及生长,甚至造成水合物的分解.对于sII型二元氢气水合物,其分解焓大小与相平衡条件、添加剂分子以及水合物中水分子间的氢键强弱等因素相关.表1 给出了不同体系下二元水合物的分解焓计算结果.从表1 可以看出,水合储氢过程中的水合物热不能忽略.氢气水合物的渐进式成核生长虽不会大幅提高体系温度,造成热抑制[40 ] .但若这些热不能及时排除,仍会使体系温度提高从而削弱水合反应速率. ...
2
... II 型二元氢气水合物分解焓[41 ] ...
... Decompound enthalpy of sII binary hydrogen hydrate[41 ] ...
1
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...
1
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...
1
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...
1
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...
2
... 对比近年来水合储氢相关实验结果(表3 ),可以发现,当水合物样品量极小时,储氢密度可以达到3%以上,而当样品量超过5 g时,储氢量就只有0.5%左右.随样品量的增加,达到100 g以上时[32 ,46 -47 ] ,储量仅剩0.1%.这些结果表明即使氢气分子进出水合物笼相较甲烷等大分子气体容易,但其储氢速率和储氢密度仍受气液接触面积以及氢气分子在水合物相中扩散的影响. ...
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
4
... 对比近年来水合储氢相关实验结果(表3 ),可以发现,当水合物样品量极小时,储氢密度可以达到3%以上,而当样品量超过5 g时,储氢量就只有0.5%左右.随样品量的增加,达到100 g以上时[32 ,46 -47 ] ,储量仅剩0.1%.这些结果表明即使氢气分子进出水合物笼相较甲烷等大分子气体容易,但其储氢速率和储氢密度仍受气液接触面积以及氢气分子在水合物相中扩散的影响. ...
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 表面活性剂是指能使溶液表面张力显著下降的物质,具有固定的亲水亲油基团,一般可分为离子型表面活性剂、非离子型表面活性剂、两性表面活性剂等.十二烷基硫酸钠(SDS)是一种典型的阴离子表面活性剂,被广泛应用于水合物领域.Linga等[47 ] 以5%摩尔分数THF为热力学促进剂,首次研究了添加不同浓度SDS的氢气水合物生成动力学.实验结果表明SDS对于提高H2 /THF二元水合物生成动力学影响甚微,水合物生成诱导时间没有出现明显降低.这与SDS促进甲烷水合物生成差异甚大;之后Veluswamy等[72 ] 以5.6%摩尔分数THF为热力学添加剂,深入研究了0.01%~1%质量分数的阳离子表面活性剂十二烷基三甲基氯化铵(DTAC)和非离子表面活性剂吐温-20(聚山梨酯20)存在下H2 /THF水合物生成动力学,研究结果表明0.5%的DTAC和0.1%的吐温-20使水合物形成速率增加约20%;随后Veluswamy等[73 ] 在搅拌釜反应器中研究了SDS对氢气/丙烷水合物动力学影响.SDS浓度在5~1000 ppm(1 ppm=0.0001%)范围内,结果表明SDS显著提高了混合水合物的形成速率,缩短了水合物的形成时间.在浓度大于100 ppm的SDS体系下,水合90%所需的时间从334.2 min减少到25.5 min.表面活性剂对氢气水合物的作用取决于客体分子和所研究的系统.对于液相促进剂,表面活性剂必须要通过亲水亲油基团形成胶束,才能达到降低表面张力,扩大气液接触面积的目的;而对于气相促进剂,SDS只需要与水形成胶束,作用机理与甲烷水合物类似,因此强化效果更加明显. ...
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
2
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 以上研究是通过分散水合物相、降低传质阻力层的方式来减少表面扩散距离,强化表面扩散通道来提高氢气水合物的形成速率.这些方法虽然在一定程度上提高了水合储氢速率,但由于热力学促进剂分子始终占据水合物大笼,也相对限制了水合储氢密度的发展.华南理工大学王燕鸿等[63 ] 提出了“沸石冰”的思路,即预先生成丙烷水合物,之后将丙烷脱除得到类沸石水合物结构;结果表明在264.3 K、10.34 MPa下“沸石冰”结构储氢无明显诱导时间,在20 min左右即完成了水合物生长过程,最终储氢容量为1.18%,同时拉曼光谱测试结果也验证了氢气分子进入了丙烷形成的水合物笼内;与丙烷-氢气混合气体水合物储氢相比,“沸石冰”储氢成核和生长时间更短,并且获得了更高的储氢容量.“沸石冰”储氢的目的在于将水合物笼中的大笼空出,为氢气分子的扩散提供了传质通道.但这种方法仅适合于气体分子充当热力学促进剂的的体系,对于液相有机促进剂无明显作用.Lee等[82 ] 在270 K、12 MPa的压力下进行了0.2%(摩尔分数,下同)、0.7%和5.6%THF溶液储氢实验,结果表明0.2%THF体系水合反应速率最快,在60 min即反应完全,5.56%THF体系水合反应速率最慢,生成时间为100 min,并且储氢量仅为1.76%,远低于0.2%THF体系所达到的3.8%储氢密度.他们将这种现象称之为“调谐效应”,即减少热力学促进剂浓度,空出部分水合物大笼,达到既可以稳定氢气水合物结构,又可以为氢气分子进入体相占据水合物大笼提供扩散通道的目的,从根本上提高了理论储氢量.Kim等[83 ] 随后基于此提出CGC(critical guest concentration)的概念,即临界客体浓度:达到最大储氢能力的液相促进剂浓度,低于此浓度可能无法使水合物结构稳定,CGC是氢气水合物调谐效应的一个重要指标.Sugahara等[48 ] 将180 μm粉状冰与固态THF相混合,制备0.54%~5.58%的固体混合物,在255 K、70 MPa下储氢,Raman光谱分析显示大笼中H2 的峰值强度随着THF摩尔分数的进一步降低而增加,这种H2 占据水合物大笼的行为与Lee等人[82 ] 报道的“调谐效应”相似.但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向. ...
3
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 除使用热力学促进剂之外,改变温度压力也是提高水合储氢性能的有效方法.表5 为不同压力下水合储氢动力学参数汇总.Trueba等[31 ] 以5.0%(摩尔分数)四氢噻吩(THT)和呋喃为促进剂,研究了二元氢气水合物与压力的关系,由表5 可知,当温度稳定在277.15 K时,压力驱动力△P 从12.0 MPa增加到28.7 MPa时,水合物生长阶段前1小时的气体吸收速率大幅提升,初始动力学速率提高了93.8%~170.0%;而t 75 随压力变化较少,储氢密度相对提高,证明提高压力可以在初始阶段维持了相对较高的生长速率,压力是促进氢气水合物成核和生长的关键因素.Ogata等[29 ] 研究了5.6%(摩尔分数)THF在11.4~66.4 MPa下水合物的储氢性能,结果表明随着压力的提高,储氢速率和储氢量明显提高,其中压力主要影响反应初期前1 h内的储氢速率,后期总体反应时间相差不大,并且随着压力的增加,储氢密度增长趋势减慢,呈现饱和状态.Sugahara等[49 ] 通过拉曼光谱、X射线衍射分析以及分解测试研究了氢气分子在THF水合物中的占据情况,结果发现即使压力提升至74 MPa,水合物储氢密度也不会显著增长,始终维持在0.7%~1%这说明提高压力增大驱动力可以提高水合储氢性能,但达到一定压力后会出现边界效应,即水合储氢性能不会出现明显改善;Burnham等[68 ] 的分子动力学模拟认为sII氢气水合物的大笼占据率很少会超过两个,在100 MPa下小笼512 的平均占据为0.85~1更符合现实,这也解释了边界效应的原因. ...
... 在275.15 K 下不同压力驱动力下水合储氢实验数据汇总表[29 , 31 , 49 ] ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
2
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 与机械搅拌等动态方式相比,静态储氢技术是一种更加节能的方式.研究表明静态冰粉强化储氢技术可以在节约能耗的同时,大幅缩短水合物成核时间,提高水合物储氢量.冰粉储氢是将促进剂(THF、CP等)水合物研磨成粉末,再与氢气进行反应.Nagai等[55 ] 以四氢呋喃为热力学促进剂,研究了温度、压力和THF颗粒尺寸对于氢气水合物生成的影响,结果表明低温、高压和小尺寸的THF颗粒在动力学上更易形成氢气水合物,同时没有明显水合物生成热效应.邓灿等[56 ] 在以环戊烷为促进剂时,对比了冰粉和溶液体系氢气水合物的形成过程.相比于搅拌溶液体系,冰粉体系的诱导时间从2.5 h降至无明显诱导时间,生成时间从30 h降至5 h左右,储氢量也相应提高了12.5%.该方法主要是通过物理粉碎降低水合物颗粒尺寸,从而增大氢气分子与水合物相之间的接触比表面. ...
2
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 与机械搅拌等动态方式相比,静态储氢技术是一种更加节能的方式.研究表明静态冰粉强化储氢技术可以在节约能耗的同时,大幅缩短水合物成核时间,提高水合物储氢量.冰粉储氢是将促进剂(THF、CP等)水合物研磨成粉末,再与氢气进行反应.Nagai等[55 ] 以四氢呋喃为热力学促进剂,研究了温度、压力和THF颗粒尺寸对于氢气水合物生成的影响,结果表明低温、高压和小尺寸的THF颗粒在动力学上更易形成氢气水合物,同时没有明显水合物生成热效应.邓灿等[56 ] 在以环戊烷为促进剂时,对比了冰粉和溶液体系氢气水合物的形成过程.相比于搅拌溶液体系,冰粉体系的诱导时间从2.5 h降至无明显诱导时间,生成时间从30 h降至5 h左右,储氢量也相应提高了12.5%.该方法主要是通过物理粉碎降低水合物颗粒尺寸,从而增大氢气分子与水合物相之间的接触比表面. ...
2
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
... 与机械搅拌等动态方式相比,静态储氢技术是一种更加节能的方式.研究表明静态冰粉强化储氢技术可以在节约能耗的同时,大幅缩短水合物成核时间,提高水合物储氢量.冰粉储氢是将促进剂(THF、CP等)水合物研磨成粉末,再与氢气进行反应.Nagai等[55 ] 以四氢呋喃为热力学促进剂,研究了温度、压力和THF颗粒尺寸对于氢气水合物生成的影响,结果表明低温、高压和小尺寸的THF颗粒在动力学上更易形成氢气水合物,同时没有明显水合物生成热效应.邓灿等[56 ] 在以环戊烷为促进剂时,对比了冰粉和溶液体系氢气水合物的形成过程.相比于搅拌溶液体系,冰粉体系的诱导时间从2.5 h降至无明显诱导时间,生成时间从30 h降至5 h左右,储氢量也相应提高了12.5%.该方法主要是通过物理粉碎降低水合物颗粒尺寸,从而增大氢气分子与水合物相之间的接触比表面. ...
1
... The effect of different sample quantities on hydrogen storage capacity
Table 3 促进剂(物质的量分数) 温度/K 压力/MPa 样品量 储氢量/% 文献 0.5%THF 255 60 1 g 3.4 Sugahara[48 ] 0.58%丙酮 255 74 1 g 3.6 Sugahara[49 ] 2.54%TBABh 253 70 1 g 0.5 Shin[50 ] 无 140 15~18 2 g 2.7 Kumar[51 ] 5.6%呋喃 275.1 41.8 3 g 0.59 Tsuda[31 ] 5.6%THT 275.1 41.5 3 g 0.6 5.56%THF 277.15 66.4 3 g 0.835 Ogata[29 ] 1.0%THF 270 13.8 5 g 0.43 Strobel[52 ] 5.56%THF 270 57 5 g 0.98 2.71%TBAB 279.5 13.8 5 g 0.214 Strobel[53 ] 5.56%THF 265.1 5.0 7.36 g 0.19 Yoshioka[54 ] 5.56%THF 266.7 6.5 10 g 0.28 Nagai[55 ] 5.6%CP 275.15 18 20 g 0.27 邓灿[56 ] 5.6%THF 270 11.6 20 g 0.4 Su[57 ] 2.0%THF 278 8.8 190 mL 0.12 Veluswamy[47 ] 3.7%TBAB 281.15 16 100 mL 0.046 Treuba[46 ] 3.4%TBAF 294.15 13 100 mL 0.024 Treuba[32 ]
水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... 水合物生成过程中气-液接触面积是影响水合物传热传质效率的关键因素.增大气-液之间的接触面积,提高体系内分子运动速率,可以增大传热传质效率,从而加快水合物生成速率;此外,气体分子与液体接触充分,也可以减少水合物空笼出现的概率,晶胞填充率得以提高,也使得储氢密度提高[58 ] .常用的增大气液接触面积的方法可分为化学方法与物理方法:化学方法主要为添加动力学促进剂,例如添加表面活性剂,通过降低溶液的表面张力,从而增大体系中的传热传质速率;物理方法有搅拌、喷雾、鼓泡等动态手段或者静态冰粉强化等技术加强传热传质速率. ...
1
... 氢气分子扩散是气相向水合物相和水合物相向气相的双向扩散,氢气分子在笼型水合物的扩散是影响水合物储氢密度的关键.分子的扩散受扩散物质的分子大小、驱动力以及扩散通道影响,通常用扩散系数来表示分子的扩散能力[59 ] . ...
1
... 此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
1
... 此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
2
... 此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
... 温度同样对水合储氢性能有明显影响,并且与压力存在显著差异.温度不仅仅影响驱动力,更与分子扩散相关.邓灿[69 ] 研究了不同温度下氢气-四氢呋喃和氢气-环戊烷体系水合储氢,结果表明储氢量和储氢速率与温度正相关,随着温度的增高而增加,并就此建立了氢气水合物扩散模型,拟合了吸附系数以及有效扩散系数.结果表明温度对吸附系数以及有效扩散系数影响明显,高温有利于氢气分子扩散;Trinh等[70 ] 通过分子动力学模拟也发现笼间氢气客体分子扩散的势垒随温度的升高而减小,温度高时氢气分子的扩散系数更大.Wang等[62 ] 的分子模拟结果也证明了,温度高时氢气分子具有更高的动能,更容易穿过水合物笼进入体相.但氢气分子的动能较高时,氢气分子也很容易从笼中逃逸;当氢气分子的动能较低时,则更容易稳定在水合物笼中,从而提高水合储氢能力.综合而言,降低温度有利于增大驱动力,但会相对降低分子扩散,因此两者之间会存在一个平衡,如何针对不同热力学促进剂体系,寻求驱动力和分子扩散的双向促进机理仍需进一步研究. ...
2
... 此外氢气的传质还受扩散通道的影响.水合物的生成是个界面反应,水合物形成后会在气液或气固界面形成水合物膜,氢气分子必须穿透表面的水合物膜才能实现进一步水合反应;随后氢气分子在水合物笼中扩散,如图4 所示.Hasegawa等[60 ] 用分子动力学模拟研究了氢气分子在sII型二元水合物中扩散的动力学过程,观察到当氢气分子在两个大笼之间扩散时,氢气分子主要通过六元环的中心扩散,扩散过程中笼结构无任何变形;但当氢气分子从一个大笼移动到小笼时,则无法通过笼的窗口进行扩散,必须要破坏部分水合物结构,即需要一个五元环断裂为氢气提供扩散通道;Gorman等[61 ] 也发现双氢气分子占据体系,在200~250 K时,氢气扩散主要是通过小笼五元环氢键的断裂和重组进行扩散;王燕鸿等[62 -63 ] 采用分子动力学模拟研究了sII型和sH型氢气二元水合物的氢气扩散行为,他们发现水合储氢过程中边界层上的512 笼将阻止氢气分子扩散到水合物相.以上研究证明氢气分子主要是通过大笼的六元环在水合物体相内进行扩散,而很难通过五元环扩散,氢气分子穿过五元环需要更大的能量来进行氢键的断裂和重组. ...
... 以上研究是通过分散水合物相、降低传质阻力层的方式来减少表面扩散距离,强化表面扩散通道来提高氢气水合物的形成速率.这些方法虽然在一定程度上提高了水合储氢速率,但由于热力学促进剂分子始终占据水合物大笼,也相对限制了水合储氢密度的发展.华南理工大学王燕鸿等[63 ] 提出了“沸石冰”的思路,即预先生成丙烷水合物,之后将丙烷脱除得到类沸石水合物结构;结果表明在264.3 K、10.34 MPa下“沸石冰”结构储氢无明显诱导时间,在20 min左右即完成了水合物生长过程,最终储氢容量为1.18%,同时拉曼光谱测试结果也验证了氢气分子进入了丙烷形成的水合物笼内;与丙烷-氢气混合气体水合物储氢相比,“沸石冰”储氢成核和生长时间更短,并且获得了更高的储氢容量.“沸石冰”储氢的目的在于将水合物笼中的大笼空出,为氢气分子的扩散提供了传质通道.但这种方法仅适合于气体分子充当热力学促进剂的的体系,对于液相有机促进剂无明显作用.Lee等[82 ] 在270 K、12 MPa的压力下进行了0.2%(摩尔分数,下同)、0.7%和5.6%THF溶液储氢实验,结果表明0.2%THF体系水合反应速率最快,在60 min即反应完全,5.56%THF体系水合反应速率最慢,生成时间为100 min,并且储氢量仅为1.76%,远低于0.2%THF体系所达到的3.8%储氢密度.他们将这种现象称之为“调谐效应”,即减少热力学促进剂浓度,空出部分水合物大笼,达到既可以稳定氢气水合物结构,又可以为氢气分子进入体相占据水合物大笼提供扩散通道的目的,从根本上提高了理论储氢量.Kim等[83 ] 随后基于此提出CGC(critical guest concentration)的概念,即临界客体浓度:达到最大储氢能力的液相促进剂浓度,低于此浓度可能无法使水合物结构稳定,CGC是氢气水合物调谐效应的一个重要指标.Sugahara等[48 ] 将180 μm粉状冰与固态THF相混合,制备0.54%~5.58%的固体混合物,在255 K、70 MPa下储氢,Raman光谱分析显示大笼中H2 的峰值强度随着THF摩尔分数的进一步降低而增加,这种H2 占据水合物大笼的行为与Lee等人[82 ] 报道的“调谐效应”相似.但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向. ...
3
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
... ,
64 -
67 ]
Phase diagram showing different promoters of the binary hydrogen hydrate<sup>[<xref ref-type="bibr" rid="R32">33</xref>, <xref ref-type="bibr" rid="R34">35</xref>, <xref ref-type="bibr" rid="R63">64</xref>-<xref ref-type="bibr" rid="R66">67</xref>]</sup> Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
... ,
64 -
67 ]
Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
1
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
1
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
3
... 图5 为已报道的sII型[33 ,64 ] 、sH型[65 -66 ] 和半笼型促进剂下[35 ,67 ] 氢气二元水合物的相平衡条件.利用不同促进剂可以形成不同结构的二元氢气水合物,对于驱动力的强化作用也不尽相同.通常来说是半笼型促进剂>sII型促进剂>sH型促进剂.表4 总结了不同晶型二元水合物储氢理论储氢密度,半笼型添加剂对二元氢气水合物的热力学促进作用最强,有利于氢气水合物成核和生长,但是半笼型结构中,可用于储存氢气的512 笼数量少,因此降低了储氢密度.综合比较分析,sII型水合物结构更适合于氢气的储存. ...
... -
67 ]
Phase diagram showing different promoters of the binary hydrogen hydrate<sup>[<xref ref-type="bibr" rid="R32">33</xref>, <xref ref-type="bibr" rid="R34">35</xref>, <xref ref-type="bibr" rid="R63">64</xref>-<xref ref-type="bibr" rid="R66">67</xref>]</sup> Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
... -
67 ]
Fig. 5 ![]()
表4 不同晶型二元水合物储氢理论储氢密度 ...
1
... 除使用热力学促进剂之外,改变温度压力也是提高水合储氢性能的有效方法.表5 为不同压力下水合储氢动力学参数汇总.Trueba等[31 ] 以5.0%(摩尔分数)四氢噻吩(THT)和呋喃为促进剂,研究了二元氢气水合物与压力的关系,由表5 可知,当温度稳定在277.15 K时,压力驱动力△P 从12.0 MPa增加到28.7 MPa时,水合物生长阶段前1小时的气体吸收速率大幅提升,初始动力学速率提高了93.8%~170.0%;而t 75 随压力变化较少,储氢密度相对提高,证明提高压力可以在初始阶段维持了相对较高的生长速率,压力是促进氢气水合物成核和生长的关键因素.Ogata等[29 ] 研究了5.6%(摩尔分数)THF在11.4~66.4 MPa下水合物的储氢性能,结果表明随着压力的提高,储氢速率和储氢量明显提高,其中压力主要影响反应初期前1 h内的储氢速率,后期总体反应时间相差不大,并且随着压力的增加,储氢密度增长趋势减慢,呈现饱和状态.Sugahara等[49 ] 通过拉曼光谱、X射线衍射分析以及分解测试研究了氢气分子在THF水合物中的占据情况,结果发现即使压力提升至74 MPa,水合物储氢密度也不会显著增长,始终维持在0.7%~1%这说明提高压力增大驱动力可以提高水合储氢性能,但达到一定压力后会出现边界效应,即水合储氢性能不会出现明显改善;Burnham等[68 ] 的分子动力学模拟认为sII氢气水合物的大笼占据率很少会超过两个,在100 MPa下小笼512 的平均占据为0.85~1更符合现实,这也解释了边界效应的原因. ...
1
... 温度同样对水合储氢性能有明显影响,并且与压力存在显著差异.温度不仅仅影响驱动力,更与分子扩散相关.邓灿[69 ] 研究了不同温度下氢气-四氢呋喃和氢气-环戊烷体系水合储氢,结果表明储氢量和储氢速率与温度正相关,随着温度的增高而增加,并就此建立了氢气水合物扩散模型,拟合了吸附系数以及有效扩散系数.结果表明温度对吸附系数以及有效扩散系数影响明显,高温有利于氢气分子扩散;Trinh等[70 ] 通过分子动力学模拟也发现笼间氢气客体分子扩散的势垒随温度的升高而减小,温度高时氢气分子的扩散系数更大.Wang等[62 ] 的分子模拟结果也证明了,温度高时氢气分子具有更高的动能,更容易穿过水合物笼进入体相.但氢气分子的动能较高时,氢气分子也很容易从笼中逃逸;当氢气分子的动能较低时,则更容易稳定在水合物笼中,从而提高水合储氢能力.综合而言,降低温度有利于增大驱动力,但会相对降低分子扩散,因此两者之间会存在一个平衡,如何针对不同热力学促进剂体系,寻求驱动力和分子扩散的双向促进机理仍需进一步研究. ...
1
... 温度同样对水合储氢性能有明显影响,并且与压力存在显著差异.温度不仅仅影响驱动力,更与分子扩散相关.邓灿[69 ] 研究了不同温度下氢气-四氢呋喃和氢气-环戊烷体系水合储氢,结果表明储氢量和储氢速率与温度正相关,随着温度的增高而增加,并就此建立了氢气水合物扩散模型,拟合了吸附系数以及有效扩散系数.结果表明温度对吸附系数以及有效扩散系数影响明显,高温有利于氢气分子扩散;Trinh等[70 ] 通过分子动力学模拟也发现笼间氢气客体分子扩散的势垒随温度的升高而减小,温度高时氢气分子的扩散系数更大.Wang等[62 ] 的分子模拟结果也证明了,温度高时氢气分子具有更高的动能,更容易穿过水合物笼进入体相.但氢气分子的动能较高时,氢气分子也很容易从笼中逃逸;当氢气分子的动能较低时,则更容易稳定在水合物笼中,从而提高水合储氢能力.综合而言,降低温度有利于增大驱动力,但会相对降低分子扩散,因此两者之间会存在一个平衡,如何针对不同热力学促进剂体系,寻求驱动力和分子扩散的双向促进机理仍需进一步研究. ...
1
... 温度同样对水合储氢性能有明显影响,并且与压力存在显著差异.温度不仅仅影响驱动力,更与分子扩散相关.邓灿[69 ] 研究了不同温度下氢气-四氢呋喃和氢气-环戊烷体系水合储氢,结果表明储氢量和储氢速率与温度正相关,随着温度的增高而增加,并就此建立了氢气水合物扩散模型,拟合了吸附系数以及有效扩散系数.结果表明温度对吸附系数以及有效扩散系数影响明显,高温有利于氢气分子扩散;Trinh等[70 ] 通过分子动力学模拟也发现笼间氢气客体分子扩散的势垒随温度的升高而减小,温度高时氢气分子的扩散系数更大.Wang等[62 ] 的分子模拟结果也证明了,温度高时氢气分子具有更高的动能,更容易穿过水合物笼进入体相.但氢气分子的动能较高时,氢气分子也很容易从笼中逃逸;当氢气分子的动能较低时,则更容易稳定在水合物笼中,从而提高水合储氢能力.综合而言,降低温度有利于增大驱动力,但会相对降低分子扩散,因此两者之间会存在一个平衡,如何针对不同热力学促进剂体系,寻求驱动力和分子扩散的双向促进机理仍需进一步研究. ...
1
... 改善气液界面通常是通过增大气液接触面积实现的,常用的方法有添加表面活性剂或机械手段、冰粉强化技术等,增强了气液两相间的传质[71 ] . ...
1
... 表面活性剂是指能使溶液表面张力显著下降的物质,具有固定的亲水亲油基团,一般可分为离子型表面活性剂、非离子型表面活性剂、两性表面活性剂等.十二烷基硫酸钠(SDS)是一种典型的阴离子表面活性剂,被广泛应用于水合物领域.Linga等[47 ] 以5%摩尔分数THF为热力学促进剂,首次研究了添加不同浓度SDS的氢气水合物生成动力学.实验结果表明SDS对于提高H2 /THF二元水合物生成动力学影响甚微,水合物生成诱导时间没有出现明显降低.这与SDS促进甲烷水合物生成差异甚大;之后Veluswamy等[72 ] 以5.6%摩尔分数THF为热力学添加剂,深入研究了0.01%~1%质量分数的阳离子表面活性剂十二烷基三甲基氯化铵(DTAC)和非离子表面活性剂吐温-20(聚山梨酯20)存在下H2 /THF水合物生成动力学,研究结果表明0.5%的DTAC和0.1%的吐温-20使水合物形成速率增加约20%;随后Veluswamy等[73 ] 在搅拌釜反应器中研究了SDS对氢气/丙烷水合物动力学影响.SDS浓度在5~1000 ppm(1 ppm=0.0001%)范围内,结果表明SDS显著提高了混合水合物的形成速率,缩短了水合物的形成时间.在浓度大于100 ppm的SDS体系下,水合90%所需的时间从334.2 min减少到25.5 min.表面活性剂对氢气水合物的作用取决于客体分子和所研究的系统.对于液相促进剂,表面活性剂必须要通过亲水亲油基团形成胶束,才能达到降低表面张力,扩大气液接触面积的目的;而对于气相促进剂,SDS只需要与水形成胶束,作用机理与甲烷水合物类似,因此强化效果更加明显. ...
1
... 表面活性剂是指能使溶液表面张力显著下降的物质,具有固定的亲水亲油基团,一般可分为离子型表面活性剂、非离子型表面活性剂、两性表面活性剂等.十二烷基硫酸钠(SDS)是一种典型的阴离子表面活性剂,被广泛应用于水合物领域.Linga等[47 ] 以5%摩尔分数THF为热力学促进剂,首次研究了添加不同浓度SDS的氢气水合物生成动力学.实验结果表明SDS对于提高H2 /THF二元水合物生成动力学影响甚微,水合物生成诱导时间没有出现明显降低.这与SDS促进甲烷水合物生成差异甚大;之后Veluswamy等[72 ] 以5.6%摩尔分数THF为热力学添加剂,深入研究了0.01%~1%质量分数的阳离子表面活性剂十二烷基三甲基氯化铵(DTAC)和非离子表面活性剂吐温-20(聚山梨酯20)存在下H2 /THF水合物生成动力学,研究结果表明0.5%的DTAC和0.1%的吐温-20使水合物形成速率增加约20%;随后Veluswamy等[73 ] 在搅拌釜反应器中研究了SDS对氢气/丙烷水合物动力学影响.SDS浓度在5~1000 ppm(1 ppm=0.0001%)范围内,结果表明SDS显著提高了混合水合物的形成速率,缩短了水合物的形成时间.在浓度大于100 ppm的SDS体系下,水合90%所需的时间从334.2 min减少到25.5 min.表面活性剂对氢气水合物的作用取决于客体分子和所研究的系统.对于液相促进剂,表面活性剂必须要通过亲水亲油基团形成胶束,才能达到降低表面张力,扩大气液接触面积的目的;而对于气相促进剂,SDS只需要与水形成胶束,作用机理与甲烷水合物类似,因此强化效果更加明显. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... 机械搅拌是水合储氢最常见的强化方式之一[31 ,74 -75 ] ,通过叶轮旋转扩大气液接触面积,同时增加流体流动性,减少界面处成核自由能,加速水合物成核,缩短水合物形成的诱导时间.由于其设计和操作简单,机械搅拌釜在水合储氢上被广泛应用.图7 对比了静态反应釜和搅拌反应器在水合储氢上的动力学性能[47 ,76 ] .结果表明机械搅拌使水合诱导时间从200 min降至2.4 min,储氢密度也相应提高了170.97%,在低驱动力6~8 MPa下搅拌反应器具有明显强化氢气水合物生成作用.鼓泡法是从反应釜底部产生气泡进入液相,使得气体更好地与液相接触.产生的气泡有效增大气-液两相的接触面积,并能增加气体在溶液中的溶解度.吕秋楠等[77 ] 研究表明鼓泡法能有效增大水合物生成速率,并且生成速率随鼓泡器气体流速的增大而增大.喷雾法[78 ] 是将水或者溶液以喷雾的形式与气体接触反应,有效增大气-液两相的传热传质效率,提高储气速率.这些动态手段都可以有效阻止水合物在界面处聚集,不断更新气液界面,对水合物的成核与生长有良好的强化效果.但随着水合物不断生成,浆液逐渐变稠,会严重影响传质效率,带来额外的能量消耗. ...
1
... Saha等[79 ] 使用多孔介质来促进氢气水合物的生成.他们以THF为促进剂,在49、65、100、226 Å四种不同孔径的活性氧化铝、硅胶、色谱柱填料中,在270 K、6.5 MPa的条件下,得到最大储氢量为1.0%;孔径为49 Å的多孔介质下四氢呋喃-氢气水合物的最短形成时间仅为27 min,生成速率是四氢呋喃水合物粉末-氢气水合物的6~22倍.水合物形成时间随多孔介质孔径的增大而增长,氢气在四氢呋喃-氢气水合物中的扩散速率随介质尺寸或者水合物粒径的增大而减小.随后Su等[80 ] 以5.56%(摩尔分数)THF为促进剂,以高吸水性的轻度交联聚丙烯酸钠盐(PSA)颗粒为载体强化水合储氢,结果发现PSA具有优异的储氢性能,仅60 min即可达到90%的储氢量.基于气体扩散通道的理念,Zhao等[81 ] 提出将氢气分子放入一维碳纳米管中的新思路,理论预测了常温常压下低维氢气水合物的形成.研究发现,与体相水合物不同,一维氢气水合物中的氢气分子形成准一维分子链被包络在一维冰纳米管内,氢气分子链可以像液体一样自由地沿轴向流动,H2 分子的轴向扩散系数要比溶液中的扩散系数大一个数量级. ...
1
... Saha等[79 ] 使用多孔介质来促进氢气水合物的生成.他们以THF为促进剂,在49、65、100、226 Å四种不同孔径的活性氧化铝、硅胶、色谱柱填料中,在270 K、6.5 MPa的条件下,得到最大储氢量为1.0%;孔径为49 Å的多孔介质下四氢呋喃-氢气水合物的最短形成时间仅为27 min,生成速率是四氢呋喃水合物粉末-氢气水合物的6~22倍.水合物形成时间随多孔介质孔径的增大而增长,氢气在四氢呋喃-氢气水合物中的扩散速率随介质尺寸或者水合物粒径的增大而减小.随后Su等[80 ] 以5.56%(摩尔分数)THF为促进剂,以高吸水性的轻度交联聚丙烯酸钠盐(PSA)颗粒为载体强化水合储氢,结果发现PSA具有优异的储氢性能,仅60 min即可达到90%的储氢量.基于气体扩散通道的理念,Zhao等[81 ] 提出将氢气分子放入一维碳纳米管中的新思路,理论预测了常温常压下低维氢气水合物的形成.研究发现,与体相水合物不同,一维氢气水合物中的氢气分子形成准一维分子链被包络在一维冰纳米管内,氢气分子链可以像液体一样自由地沿轴向流动,H2 分子的轴向扩散系数要比溶液中的扩散系数大一个数量级. ...
1
... Saha等[79 ] 使用多孔介质来促进氢气水合物的生成.他们以THF为促进剂,在49、65、100、226 Å四种不同孔径的活性氧化铝、硅胶、色谱柱填料中,在270 K、6.5 MPa的条件下,得到最大储氢量为1.0%;孔径为49 Å的多孔介质下四氢呋喃-氢气水合物的最短形成时间仅为27 min,生成速率是四氢呋喃水合物粉末-氢气水合物的6~22倍.水合物形成时间随多孔介质孔径的增大而增长,氢气在四氢呋喃-氢气水合物中的扩散速率随介质尺寸或者水合物粒径的增大而减小.随后Su等[80 ] 以5.56%(摩尔分数)THF为促进剂,以高吸水性的轻度交联聚丙烯酸钠盐(PSA)颗粒为载体强化水合储氢,结果发现PSA具有优异的储氢性能,仅60 min即可达到90%的储氢量.基于气体扩散通道的理念,Zhao等[81 ] 提出将氢气分子放入一维碳纳米管中的新思路,理论预测了常温常压下低维氢气水合物的形成.研究发现,与体相水合物不同,一维氢气水合物中的氢气分子形成准一维分子链被包络在一维冰纳米管内,氢气分子链可以像液体一样自由地沿轴向流动,H2 分子的轴向扩散系数要比溶液中的扩散系数大一个数量级. ...
2
... 以上研究是通过分散水合物相、降低传质阻力层的方式来减少表面扩散距离,强化表面扩散通道来提高氢气水合物的形成速率.这些方法虽然在一定程度上提高了水合储氢速率,但由于热力学促进剂分子始终占据水合物大笼,也相对限制了水合储氢密度的发展.华南理工大学王燕鸿等[63 ] 提出了“沸石冰”的思路,即预先生成丙烷水合物,之后将丙烷脱除得到类沸石水合物结构;结果表明在264.3 K、10.34 MPa下“沸石冰”结构储氢无明显诱导时间,在20 min左右即完成了水合物生长过程,最终储氢容量为1.18%,同时拉曼光谱测试结果也验证了氢气分子进入了丙烷形成的水合物笼内;与丙烷-氢气混合气体水合物储氢相比,“沸石冰”储氢成核和生长时间更短,并且获得了更高的储氢容量.“沸石冰”储氢的目的在于将水合物笼中的大笼空出,为氢气分子的扩散提供了传质通道.但这种方法仅适合于气体分子充当热力学促进剂的的体系,对于液相有机促进剂无明显作用.Lee等[82 ] 在270 K、12 MPa的压力下进行了0.2%(摩尔分数,下同)、0.7%和5.6%THF溶液储氢实验,结果表明0.2%THF体系水合反应速率最快,在60 min即反应完全,5.56%THF体系水合反应速率最慢,生成时间为100 min,并且储氢量仅为1.76%,远低于0.2%THF体系所达到的3.8%储氢密度.他们将这种现象称之为“调谐效应”,即减少热力学促进剂浓度,空出部分水合物大笼,达到既可以稳定氢气水合物结构,又可以为氢气分子进入体相占据水合物大笼提供扩散通道的目的,从根本上提高了理论储氢量.Kim等[83 ] 随后基于此提出CGC(critical guest concentration)的概念,即临界客体浓度:达到最大储氢能力的液相促进剂浓度,低于此浓度可能无法使水合物结构稳定,CGC是氢气水合物调谐效应的一个重要指标.Sugahara等[48 ] 将180 μm粉状冰与固态THF相混合,制备0.54%~5.58%的固体混合物,在255 K、70 MPa下储氢,Raman光谱分析显示大笼中H2 的峰值强度随着THF摩尔分数的进一步降低而增加,这种H2 占据水合物大笼的行为与Lee等人[82 ] 报道的“调谐效应”相似.但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向. ...
... [82 ]报道的“调谐效应”相似.但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向. ...
1
... 以上研究是通过分散水合物相、降低传质阻力层的方式来减少表面扩散距离,强化表面扩散通道来提高氢气水合物的形成速率.这些方法虽然在一定程度上提高了水合储氢速率,但由于热力学促进剂分子始终占据水合物大笼,也相对限制了水合储氢密度的发展.华南理工大学王燕鸿等[63 ] 提出了“沸石冰”的思路,即预先生成丙烷水合物,之后将丙烷脱除得到类沸石水合物结构;结果表明在264.3 K、10.34 MPa下“沸石冰”结构储氢无明显诱导时间,在20 min左右即完成了水合物生长过程,最终储氢容量为1.18%,同时拉曼光谱测试结果也验证了氢气分子进入了丙烷形成的水合物笼内;与丙烷-氢气混合气体水合物储氢相比,“沸石冰”储氢成核和生长时间更短,并且获得了更高的储氢容量.“沸石冰”储氢的目的在于将水合物笼中的大笼空出,为氢气分子的扩散提供了传质通道.但这种方法仅适合于气体分子充当热力学促进剂的的体系,对于液相有机促进剂无明显作用.Lee等[82 ] 在270 K、12 MPa的压力下进行了0.2%(摩尔分数,下同)、0.7%和5.6%THF溶液储氢实验,结果表明0.2%THF体系水合反应速率最快,在60 min即反应完全,5.56%THF体系水合反应速率最慢,生成时间为100 min,并且储氢量仅为1.76%,远低于0.2%THF体系所达到的3.8%储氢密度.他们将这种现象称之为“调谐效应”,即减少热力学促进剂浓度,空出部分水合物大笼,达到既可以稳定氢气水合物结构,又可以为氢气分子进入体相占据水合物大笼提供扩散通道的目的,从根本上提高了理论储氢量.Kim等[83 ] 随后基于此提出CGC(critical guest concentration)的概念,即临界客体浓度:达到最大储氢能力的液相促进剂浓度,低于此浓度可能无法使水合物结构稳定,CGC是氢气水合物调谐效应的一个重要指标.Sugahara等[48 ] 将180 μm粉状冰与固态THF相混合,制备0.54%~5.58%的固体混合物,在255 K、70 MPa下储氢,Raman光谱分析显示大笼中H2 的峰值强度随着THF摩尔分数的进一步降低而增加,这种H2 占据水合物大笼的行为与Lee等人[82 ] 报道的“调谐效应”相似.但目前关于调谐效应仍存在争议,如何使调谐效应更好地应用是今后需要重点关注的方向. ...
1
... 在传质方面,最初认为由于氢气分子尺寸原因,氢气水合物的生长不存在传质阻碍[42 ] .如表2 所示,氢气分子的分子直径仅为2.3 Å(1 Å=0.1 nm),远小于甲烷、二氧化碳等气体分子直径和水合物笼直径(氢气分子与水合物孔穴直径比仅有0.3~0.5),并且氢气分子逸度很大,可以自由进出水合物笼,这也是氢气水合物难以稳定的原因之一[8 ] .中国石油大学(北京)陈光进教授团队等[43 ] 用拉曼光谱研究H2 、H2 +CH4 和H2 +CO2 在THF水合物生长和迁移规律,结果表明H2 分子可以在5 mm厚的THF水合物层内进行渗透、迁移和储存,其中H2 分子在THF水合物中渗透迁移所需要的最小分压为2.9 MPa,其扩散系数为6.1×10-12 m2 /s,远大于CH4 (9.4×10-17 m2 /s)[44 ] 和CO2 (约10-18 m2 /s)[45 ] 的扩散系数,表明了水合物层对CH4 和CO2 具有明显传质阻力;Guillermo等[84 ] 使用从头算密度泛函理论研究了水合物中CH4 、CO2 和H2 分子的吸附和扩散,扩散活化能分别为1.0、0.4和0.2 eV,种种研究表明H2 分子只是比CH4 、CO2 等气体分子更容易在水合物层扩散,但是仍需要一定驱动力,水合储氢过程仍需考虑传质问题. ...




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}