Review of the molecular dynamics of molten salt thermal physical properties
FU Dianwei,, ZHANG Cancan,, NA Heya, WANG Guoqiang, WU Yuting, LU Yuanwei
MOE Key Laboratory of Enhanced Heat Transfer and Energy Conservation, BeijingKey Laboratory of Heat Transfer and Energy Conversion, College of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124, China
As a high-temperature heat transfer and storage medium, molten salt is widely used for solar thermal power generation and the flexible transformation of thermal power plants. First, the potential functions of the molecular dynamics of molten salt were summarized and analyzed. This indicated that to reduce simulation errors, the Buckingham potential with coulomb force is more suitable for nitrate and the BMH potential is more suitable for carbonate and chloride salt. Second, an analysis of the thermal properties of molten salt indicated that the addition of Ca2+ to solar salt decreased its melting point and increased its viscosity, and the specific heat capacity of nitrate decreased with increasing NO2- concentration. Increased Li+ concentrations increased the specific heat capacity and thermal conductivity of chloride salt but also increased the simulation error; however, with increased K+, the specific heat capacity error decreased and the error when calculating residual heat properties increased. The carbonate simulation error was relatively small, which is consistent with experimental results. The simulation errors were large with the addition of K+ or Li+, and the increased potential energy between ions led to the loss of some particles. It was found that the influence of the boundary effect after the introduction of a boundary condition increased the error; however, the error was reduced by increasing the number of molecules, the potential energy truncation distance correction, and the simulation time step. Currently, studies on the molecular dynamics of molten salt with the same cation and different anions are rare. Exploring the influence of nanofluids on molten salt molecular dynamics, reducing the simulation error of molecular dynamics, and conducting research on the corrosion characteristics of molten salt based on molecular dynamics is the next research direction of molten salt molecular dynamics.
Keywords:molten salt
;
molecular dynamics
;
potential function
;
thermophysical property
FU Dianwei. Review of the molecular dynamics of molten salt thermal physical properties[J]. Energy Storage Science and Technology, 2023, 12(12): 3873-3882
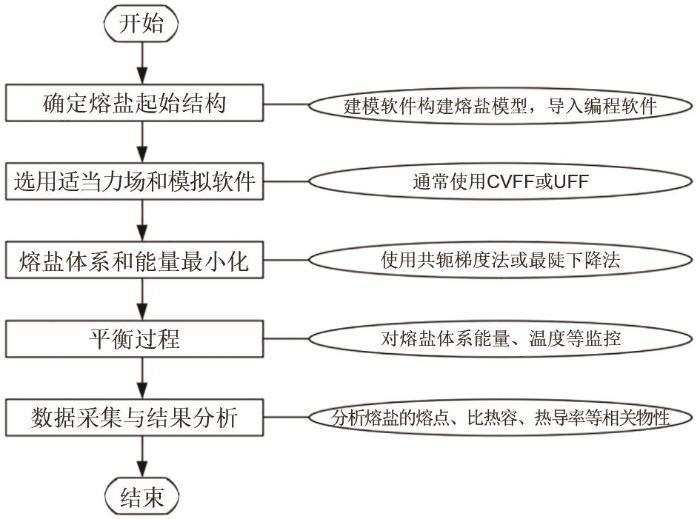
Fig. 2
The flow chart of molten salt molecular dynamics
1.1 势函数的研究进展
分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础。
分子动力学模拟通常选择CVFF (consistent valence force field)力场或者UFF (universal force field)力场。CVFF力场属于传统力场,适用于有机小分子体系,主要预测分子结构和自由能;UFF力场通常适用于所有元素。早期的原子间相互作用势,大多是一些纯经验拟合势,近年来通过对基本电子结构的理论计算,根据对系统总能量的贡献将其分为两类:①总能量由势函数决定,可有效描述范德瓦耳斯互作用占据主导地位的体系;②势函数仅仅描述恒定材料平均密度下系统的能量随原子构型的变化,适用于描述sp-价态金属。本文对熔盐常用的势函数类型、计算公式及对应熔盐进行了总结,如表1所示。
Table 1
表1
表1熔盐分子动力学模拟势函数
Table 1 Molten salt molecular dynamic simulation potential functions
分子动力学计算中用到多种公式,决定分子间的相互作用力。常用的一些公式包括径向分布函数(radial distribution function,RDF)、配位数N(r)、自扩散系数D、均方位移(mean square displacement,MSD)、角分布函数(angular distribution function,ADF)、热导率等,如表2所示。
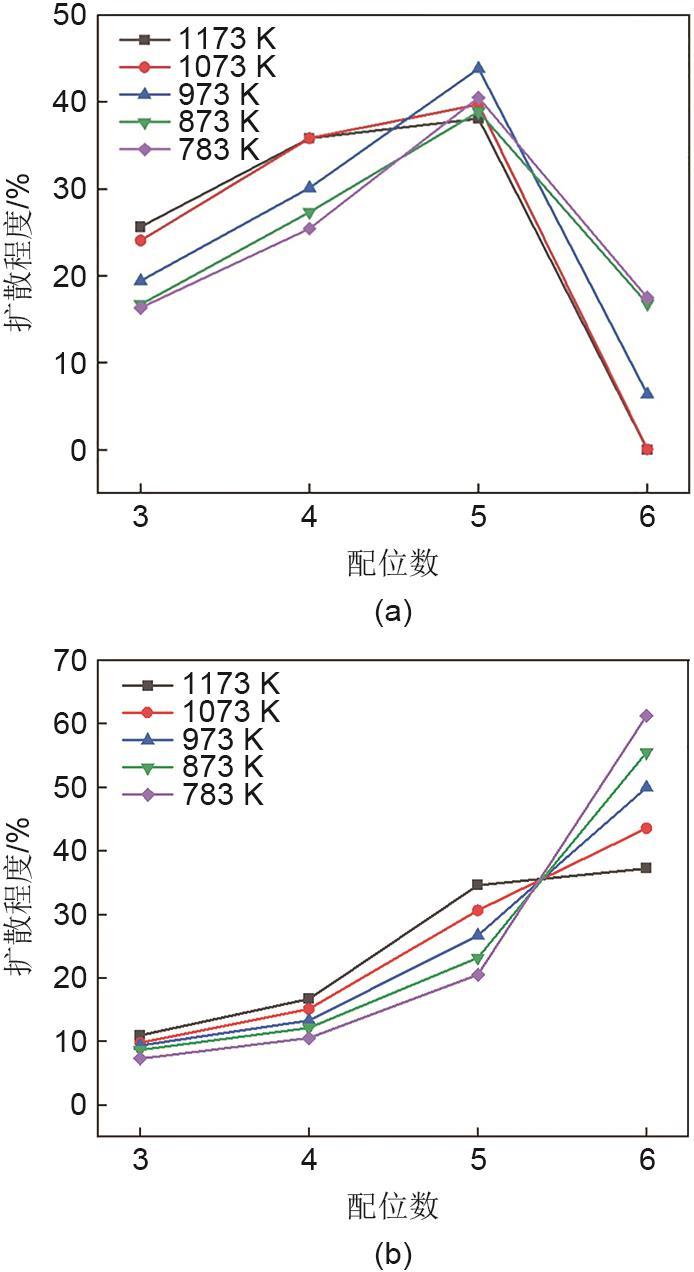
Fig. 3
The variation of diffusion degree with temperature and coordination number of , ion pairs and Ca2+, ion pairs: (a) , ion pair; (b) Ca2+, ion pair
WANG H X, XIONG Y X, REN J, et al. Fabrication and performance investigation of Na2CO3/Carbide slag shape-stable phase change composites[J]. Energy Storage Science and Technology, 2022, 11(12): 3819-3827.
LIU Z C, PENG D G, ZHAO H R, et al. Development prospects of energy storage participating in auxiliary services of power systems under the targets of the dual-carbon goal[J]. Energy Storage Science and Technology, 2022, 11(2): 704-716.
ZHANG L D, WU Y T, REN N, et al. Effects of nanoparticle dispersion on enhancing specific heat capacity of lmps salt[J]. Acta Energiae Solaris Sinica, 2017, 38(11): 3018-3021.
WANG J L, WU H D. Thermophysical properties of solar salt and its application in solar thermal power generation[J]. Power Equipment, 2021, 35(5): 334-338.
XU B, LI P W, CHAN C. Application of phase change materials for thermal energy storage in concentrated solar thermal power plants: A review to recent developments[J]. Applied Energy, 2015, 160: 286-307.
ARAMENDı́A M A, BORAU V, JIMÉNEZ C, et al. Effects of Na2CO3 impregnation on the catalytic activity of Mg3(PO4)2 in the gas-phase conversion of 2-hexanol and the alkylation of aniline with methanol[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2000, 170(1): 51-58.
BAE S J, AHN Y, LEE J, et al. Various supercritical carbon dioxide cycle layouts study for molten carbonate fuel cell application[J]. Journal of Power Sources, 2014, 270: 608-618.
IORA P, CAMPANARI S. Development of a three-dimensional molten carbonate fuel cell model and application to hybrid cycle simulations[J]. Journal of Fuel Cell Science and Technology, 2007, 4(4): 501-510.
LIAO M, DING J, WEI X L, et al. Preparation and heat transfer and thermal storage property of high-temperature carbonate molten salt[J]. Inorganic Chemicals Industry, 2008, 40(10): 15-17.
SUBARI F, MAKSOM H F, ZAWAWI A. Corrosion behavior of eutectic molten salt solution on stainless steel 316L[J]. Procedia-Social and Behavioral Sciences, 2015, 195: 2699-2708.
TAFRISHI H, SADEGHZADEH S, AHMADI R. Molecular dynamics simulations of phase change materials for thermal energy storage: A review[J]. RSC Advances, 2022, 12(23): 14776-14807.
CUI S X, HU H Q, XIAO X G, et al. The basic principles and methods of molecular dynamics simulation[J]. Journal of Liaocheng Teachers University, 2005, 18(1): 30-34.
DIENES G, HATCHER R, SMOLCHOWSKI R, et al. Recent calculations concerning point defects in alkali halides[J]. Miscellaneous Publication-National Bureau of Standards, 1934: 11.
BUCKINGHAM R A. The classical equation of state of gaseous helium, neon and argon[J]. Proceedings of the Royal Society of London Series A Mathematical and Physical Sciences, 1938, 168(933): 264-283.
GALAMBA N, NIETO DE CASTRO C A, ELY J F. Thermal conductivity of molten alkali halides from equilibrium molecular dynamics simulations[J]. The Journal of Chemical Physics, 2004, 120(18): 8676-8682.
WU J, WANG J, NI H O, et al. The influence of NaCl concentration on the (LiCl-KCl)eutectic system and temperature dependence of the ternary system[J]. Journal of Molecular Liquids, 2018, 253: 96-112.
ZHANG S, YAN Y Y. Melting and thermodynamic properties of nanoscale binary chloride salt as high-temperature energy storage material[J]. Case Studies in Thermal Engineering, 2021, 25: 100973.
FUMI F G, TOSI M P. Ionic sizes and born repulsive parameters in the NaCl-type alkali halides—I[J]. Journal of Physics and Chemistry of Solids, 1964, 25(1): 31-43.
TOSI M P, FUMI F G. Ionic sizes and born repulsive parameters in the NaCl-type alkali halides—II[J]. Journal of Physics and Chemistry of Solids, 1964, 25(1): 45-52.
SUN H. COMPASS: An ab initio force-field optimized for condensed-phase ApplicationsOverview with details on alkane and benzene compounds[J]. Journal of Physical Chemistry B, 1998, 102: 7338-7364.
WHITE D N J. A computationally efficient alternative to the Buckingham potential for molecular mechanics calculations[J]. Journal of Computer-Aided Molecular Design, 1997, 11(5): 517-521.
HU Y W, HE Y R, ZHANG Z D, et al. Effect of Al2O3 nanoparticle dispersion on the specific heat capacity of a eutectic binary nitrate salt for solar power applications[J]. Energy Conversion and Management, 2017, 142: 366-373.
PAN G C, DING J, WANG W L, et al. Molecular simulations of the thermal and transport properties of alkali chloride salts for high-temperature thermal energy storage[J]. International Journal of Heat and Mass Transfer, 2016, 103: 417-427.
NI H O, WU J, SUN Z, et al. Molecular simulation of the structure and physical properties of alkali nitrate salts for thermal energy storage[J]. Renewable Energy, 2019, 136: 955-967.
HAZEBROUCQ S, PICARD G S, ADAMO C, et al. Density-functional-based molecular-dynamics simulations of molten salts[J]. The Journal of Chemical Physics, 2005, 123(13): 134510.
DING J, PAN G, DU L C, et al. Theoretical prediction of the local structures and transport properties of binary alkali chloride salts for concentrating solar power[J]. Nano Energy, 2017, 39: 380-389.
XIE W J, DING J, PAN G, et al. Heat and mass transportation properties of binary chloride salt as a high-temperature heat storage and transfer media[J]. Solar Energy Materials and Solar Cells, 2020, 209: 110415.
RONG Z Z, PAN G, LU J F, et al. Ab-initio molecular dynamics study on thermal property of NaCl-CaCl2 molten salt for high-temperature heat transfer and storage[J]. Renewable Energy, 2021, 163: 579-588.
JIANG T, WANG N, CHENG C M, et al. Molecular dynamics simulation on the structure and thermodynamics of molten LiCl-KCl-CeCl3[J]. Acta Physico-Chimica Sinica, 2016, 32(3): 647-655.
ANAGNOSTOPOULOS A, ALEXIADIS A, DING Y. Molecular dynamics simulation of solar salt (NaNO3-KNO3) mixtures[J]. Solar Energy Materials and Solar Cells, 2019, 200: 109897.
JAYARAMAN S, THOMPSON A, LILIENFELD O A V, et al. Molecular simulation of the thermal and transport properties of three alkali nitrate salts[J]. Industrial \& Engineering Chemistry Research, 2010, 49: 559-571.
WU Y T, REN N, LIU B, et al. Heat transfer and heat storage of molten salt and its application in solar thermal power generation[J]. Advanced Materials Industry, 2012(7): 20-26.
ZHANG C C, WU Y T, LU Y W. Preparation and comparative analysis of thermophysical properties on low melting point mixed nitrate molten salts[J]. Energy Storage Science and Technology, 2020, 9(2): 435-439.
WEI G S, WANG G, XU C, et al. Selection principles and thermophysical properties of high temperature phase change materials for thermal energy storage: A review[J]. Renewable and Sustainable Energy Reviews, 2018, 81: 1771-1786.
HE Z Y, YANG Q R, LI Z Y, et al. Effect of the mesoporous size, structure and surface on the melting and heat transport properties of solar salt[J]. Solar Energy Materials and Solar Cells, 2022, 248: 111978.
NI H O, WU J, SUN Z, et al. Insight into the viscosity enhancement ability of Ca(NO3)2 on the binary molten nitrate salt: A molecular dynamics simulation study[J]. Chemical Engineering Journal, 2019, 377: 120029.
CHEN Y Y, ZHAO C Y. Thermophysical properties of Ca(NO3)2-NaNO3-KNO3 mixtures for heat transfer and thermal storage[J]. Solar Energy, 2017, 146: 172-179.
NI H O, WU J, SUN Z, et al. Influence of NO2-on the microscopic structure and physical properties of the binary nitrate salts: A molecular dynamics simulation study[J]. Journal of Thermal Science, 2020, 29(2): 464-476.
MAHTAB A F, DONGHYUN S. Molecular dynamics study on the impact of the development of dendritic nanostructures on the specific heat capacity of molten salt nanofluids[J]. Journal of Energy Storage, 2023, 71: 107850.
YANG X M, LIU J T, CUI J X, et al. Molecular dynamics simulation research of thermophysical properties of chloride molten salts and their mixtures[J]. Acta Energiae Solaris Sinica, 2022, 43(11): 433-442.
DING J, PAN G, DU L C, et al. Molecular dynamics simulations of the local structures and transport properties of Na2CO3 and K2CO3[J]. Applied Energy, 2018, 227: 555-563.
MANGA V R, SWINTECK N, BRINGUIER S, et al. Interplay between structure and transport properties of molten salt mixtures of ZnCl2-NaCl-KCl: A molecular dynamics study[J]. The Journal of Chemical Physics, 2016, 144(9): 094501.
RUAN Z H, GAO P, YUAN Y, et al. Theoretical estimation of temperature-dependent radiation properties of molten solar salt using molecular dynamics and first principles[J]. Energy, 2022, 246: 123379.
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
1
... 分子动力学模拟结果准确性与系统内原子间相互作用势的选取存在直接关系[17],1924—1984年,学者先后提出了Lennard-Jones(LJ)[18]、Morse[19]、Born-Mayer[20]、Buckingham[21]、Busing[22]和Embedded Atom Method(EAM)[23]势函数解析式,为熔盐分子动力学研究奠定了基础. ...
2
... Molten salt molecular dynamic simulation potential functionsTable 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}