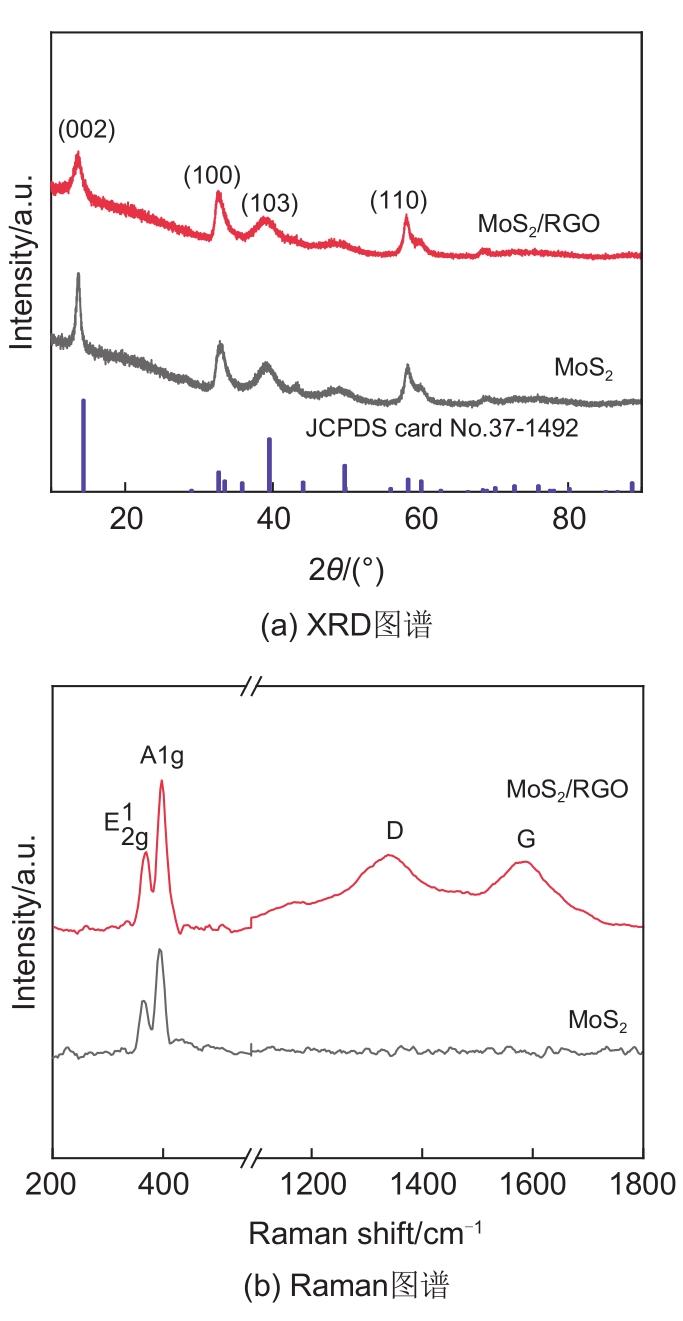
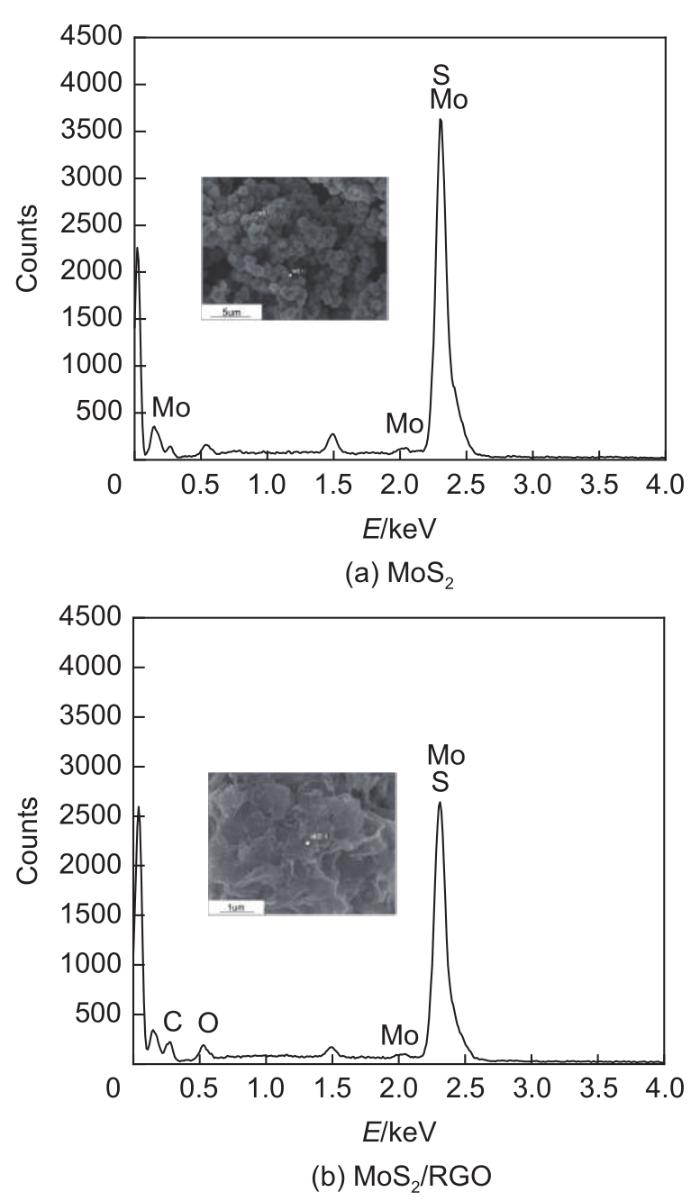
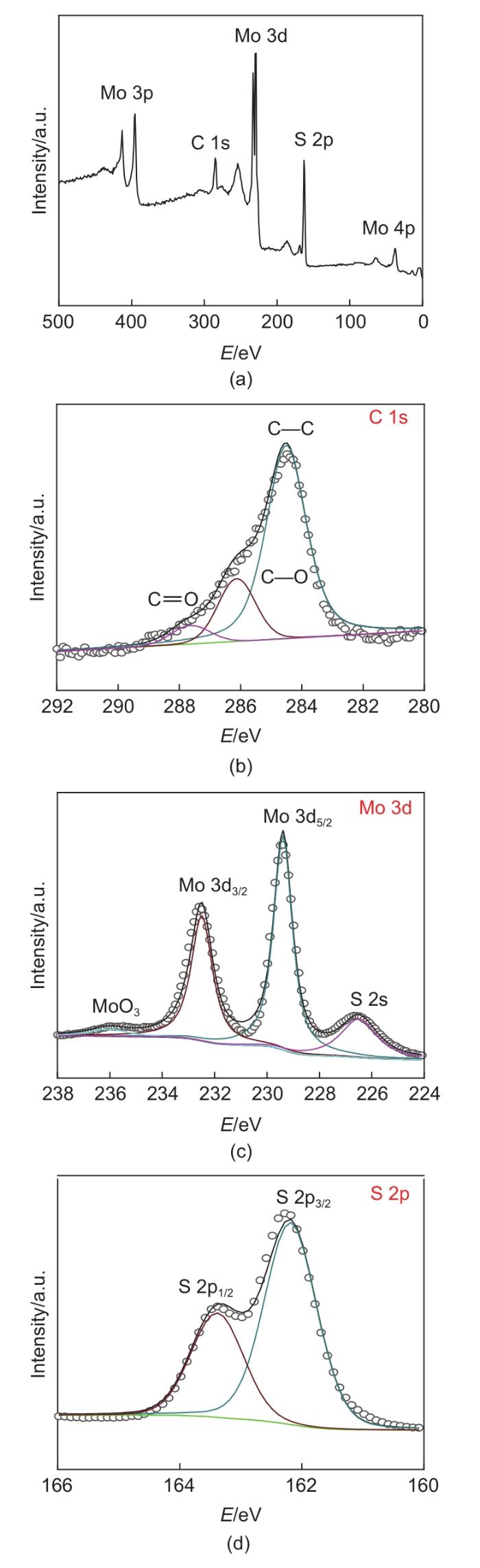
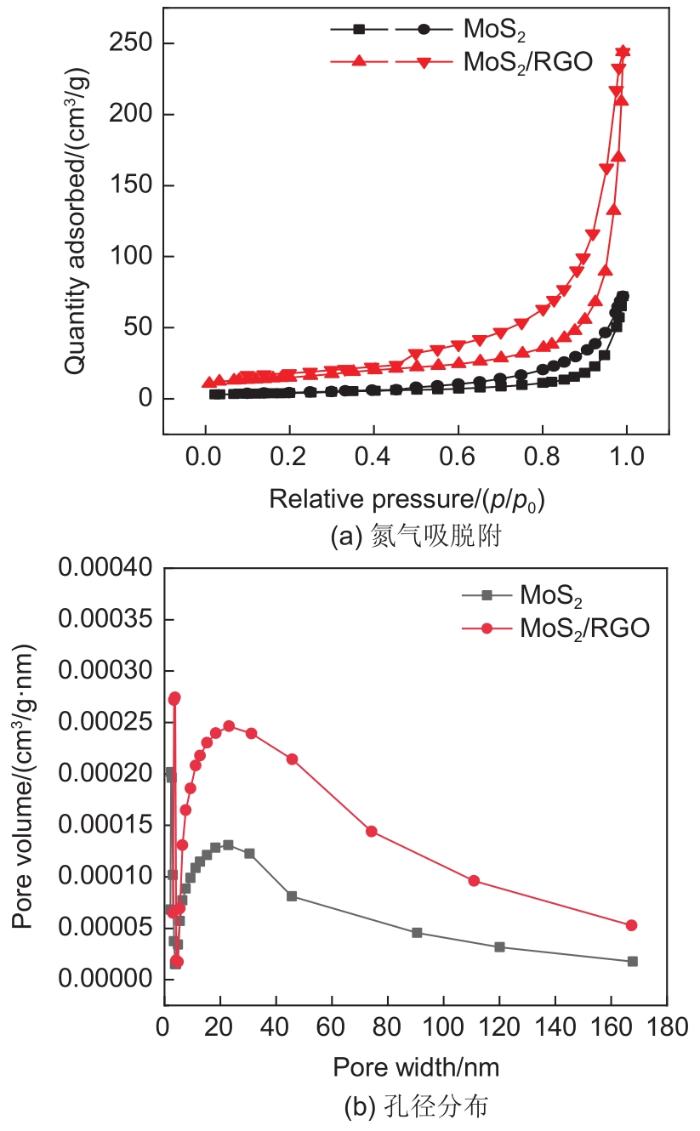
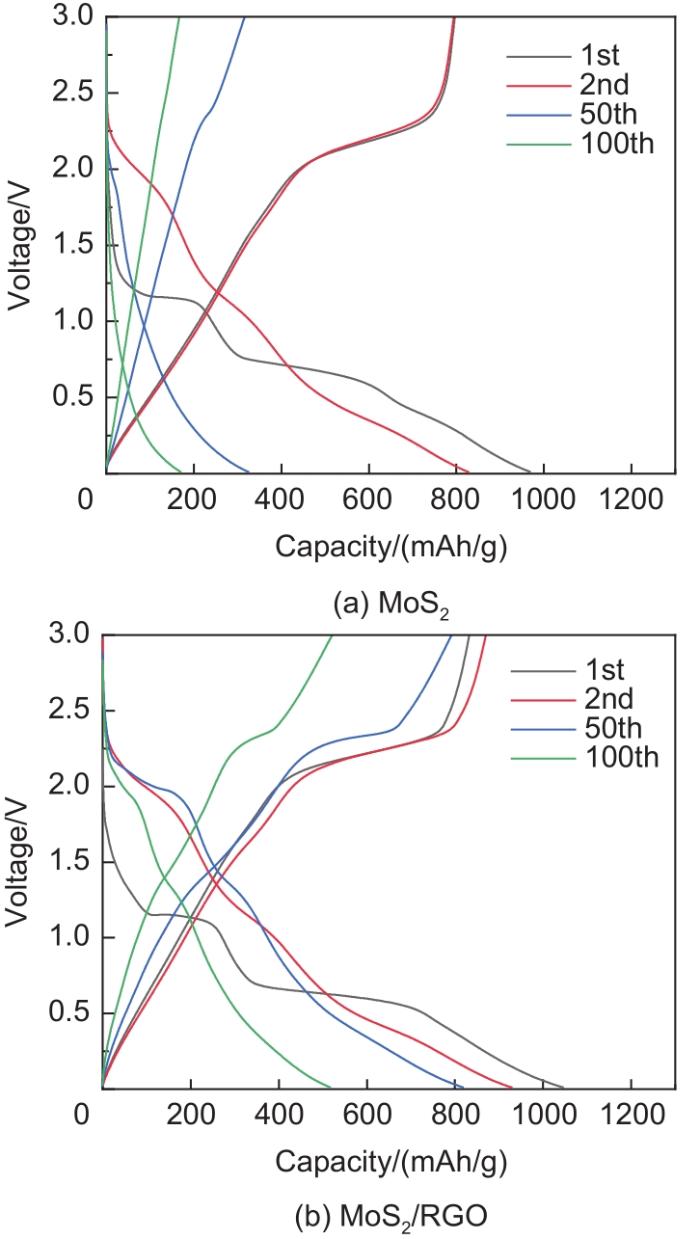
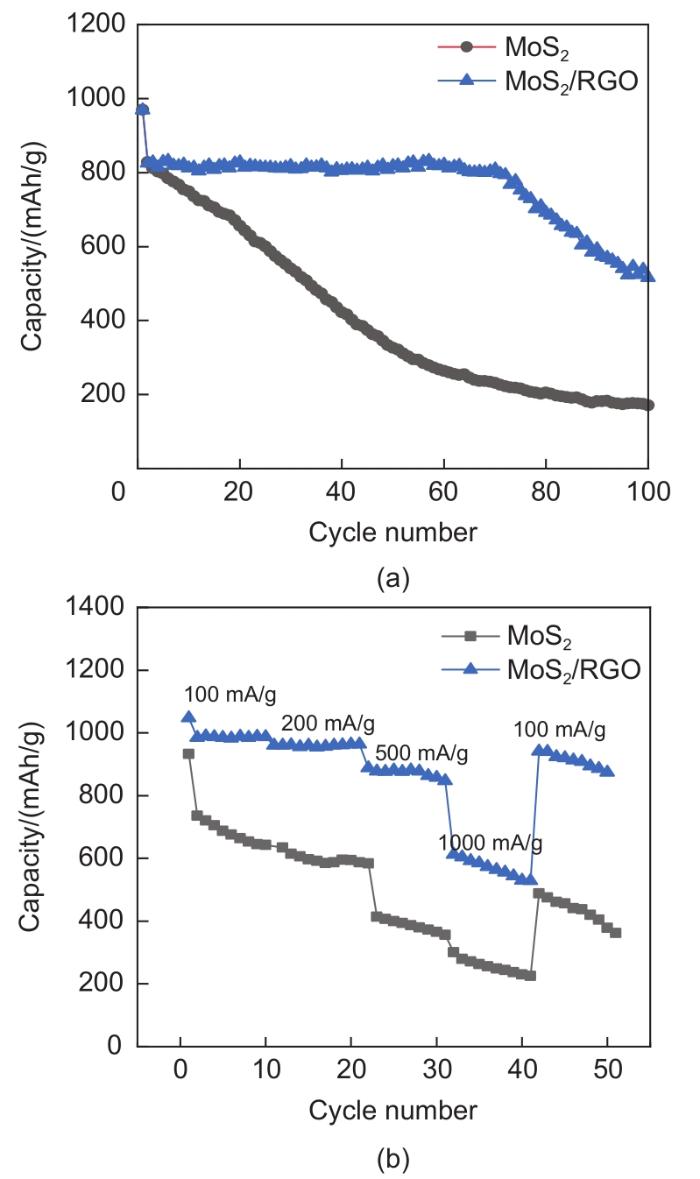
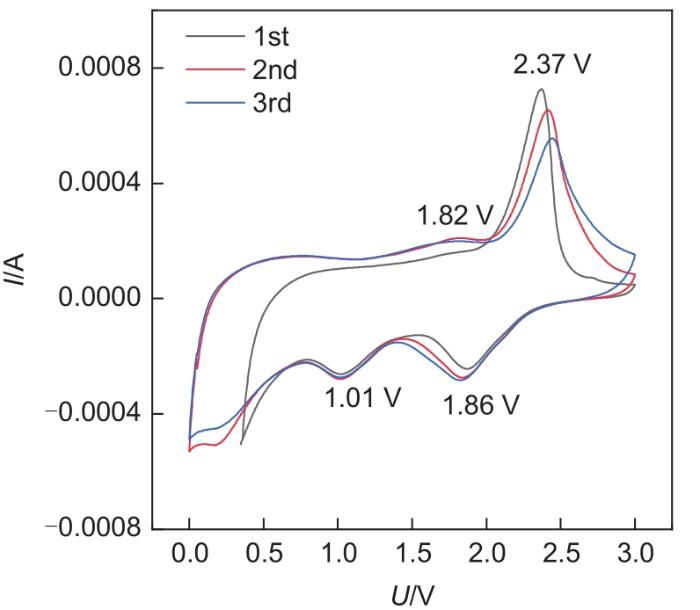
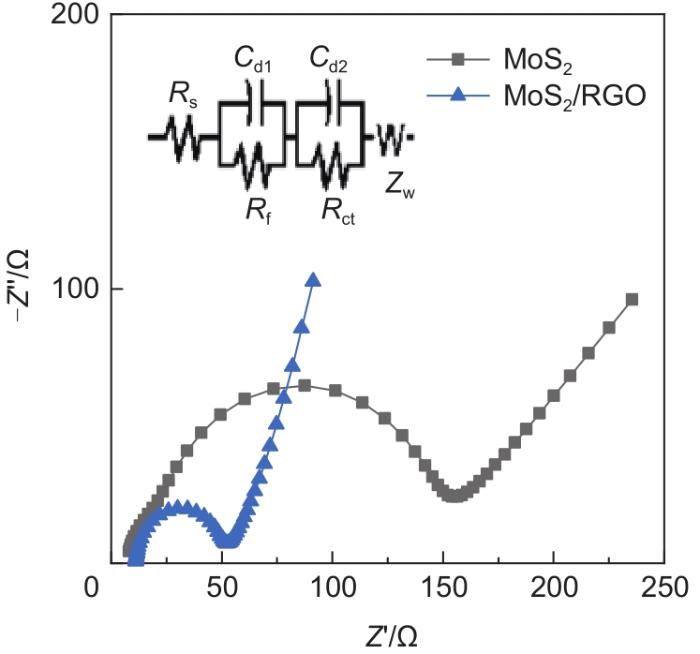
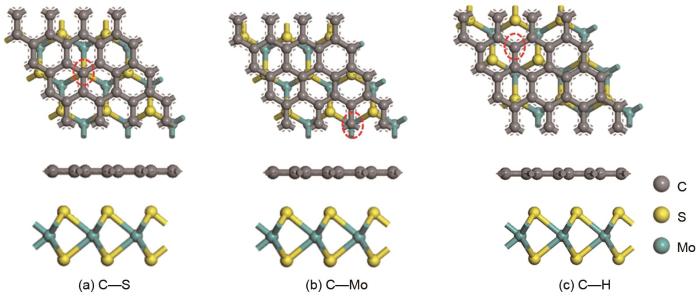
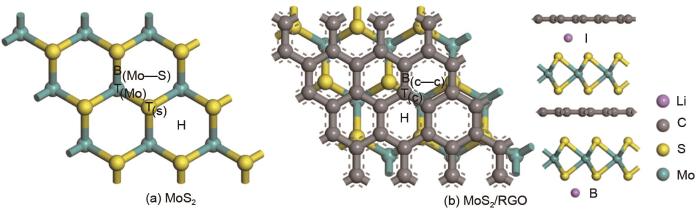
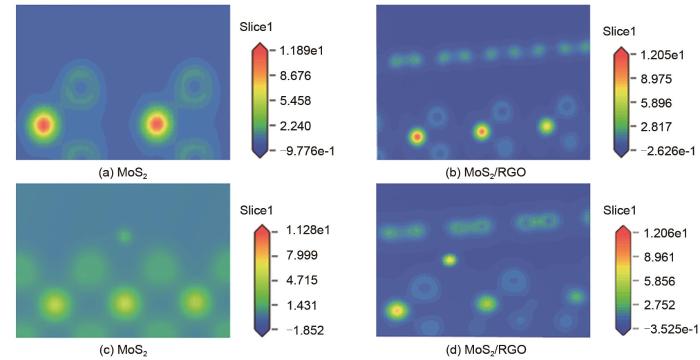
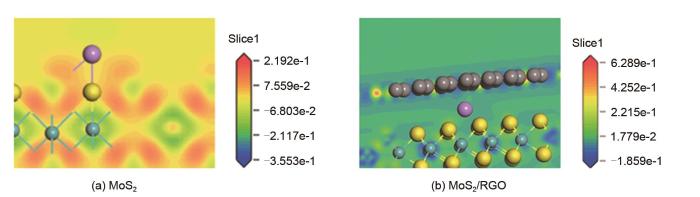
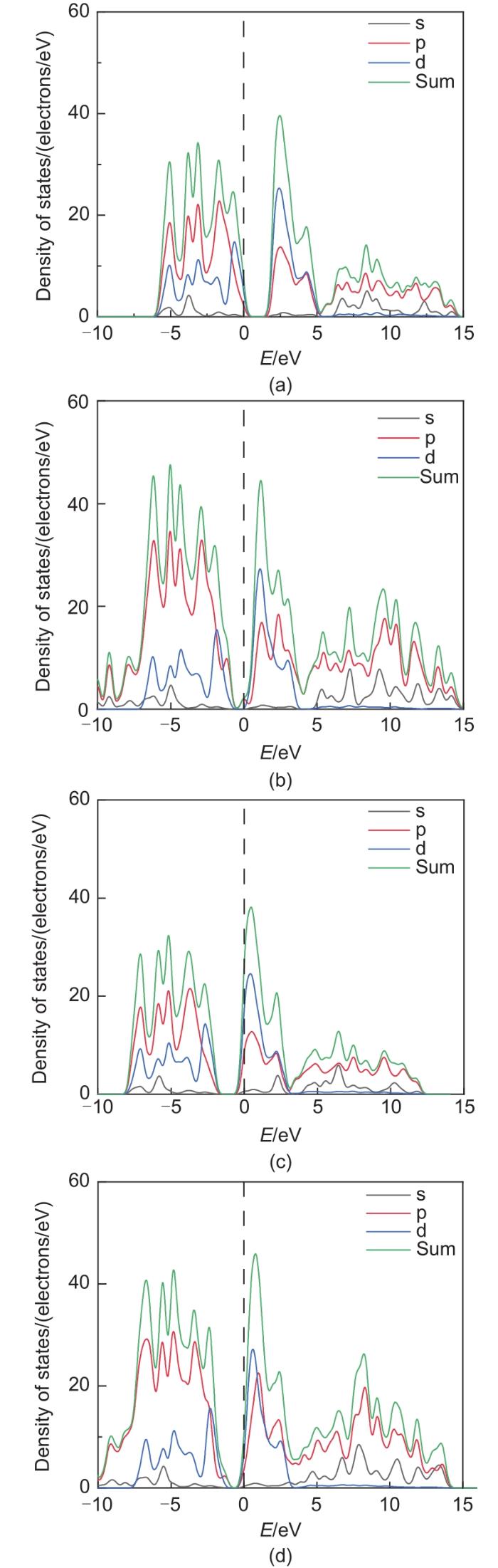
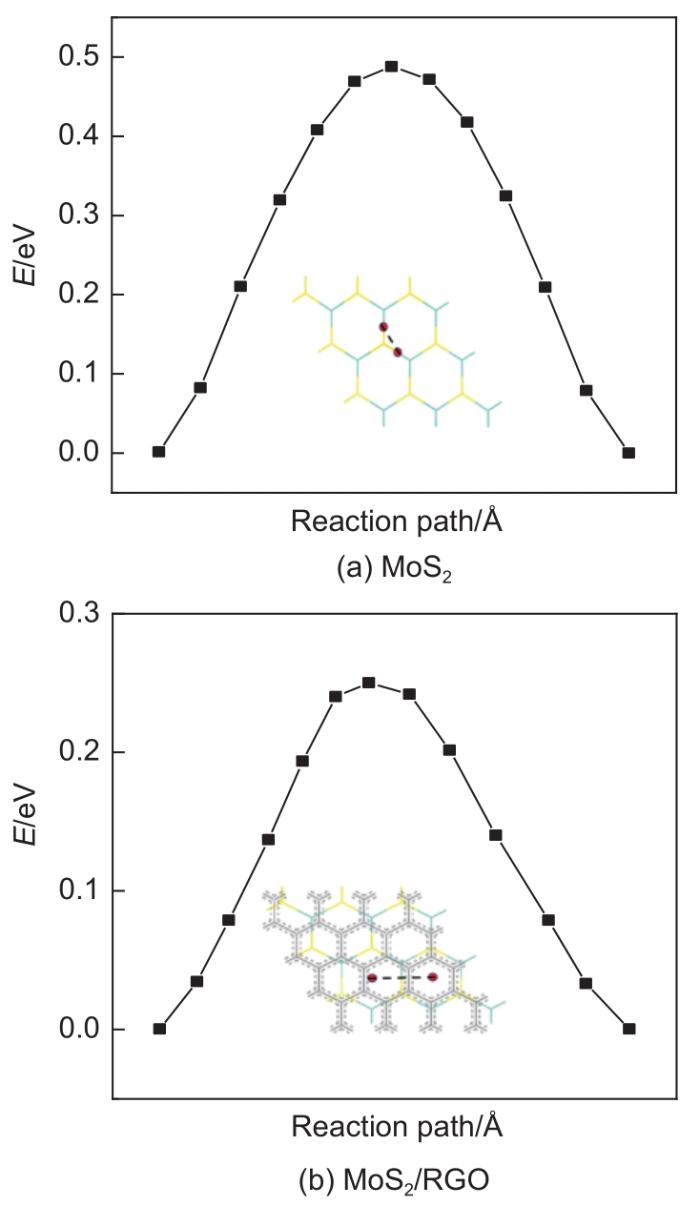
Based on the high theoretical lithium storage capacity of molybdenum disulfide (MoS2) and the good conductivity of graphene, a curly lamellar MoS2/reduced graphene oxide (RGO) composite was successfully prepared by a one-step hydrothermal method. The structure, morphology, and composition of the MoS2/RGO composite were characterized using an X-ray diffractometer, scanning electron microscope, X-ray energy spectrometer, and Raman spectrometer. The most stable adsorption position of lithium-ion, charge density, charge density difference, density of states, and diffusion energy barrier of MoS2 and MoS2/RGO models were calculated by first-principles. Results show that the MoS2/RGO composite maintains a high discharge specific capacity of more than 800 mAh/g in the first 70 charge and discharge cycles. After 100 cycles, the discharge specific capacity of the MoS2/RGO composite is 515.3 mAh/g, which is significantly higher than that of MoS2 (170.8 mAh/g). Simultaneously, the composite material shows a better rate of performance than MoS2. When the current density is back to 100 mA/g after the 1000 mA/g high current density cycle, the MoS2/RGO composite still maintains a high discharge specific capacity (941.2 mAh/g). The first-principles calculation results show that the charge near the Mo atom of MoS2 decreases, and the whole density of states of the MoS2/RGO composite is enhanced due to the action of graphene, making it easier for electrons in the valence band to migrate to the conduction band. Furthermore, compared with MoS2, the low diffusion energy barrier (0.25 eV) of MoS2/RGO makes it easier for lithium ions to diffuse. Therefore, it explains why the MoS2/RGO composite has a better electrochemical performance than MoS2 with the effect of graphene.
ZHU Yuting. Electrochemical properties and First-principles study of MoS2/rGO composite[J]. Energy Storage Science and Technology, 2023, 12(3): 698-709
FAIZAN M, HUSSAIN S, VIKRAMAN D, et al. MoS2@Mo2C hybrid nanostructures formation as an efficient anode material for lithium-ion batteries[J]. Journal of Materials Research and Technology, 2021, 14: 2382-2393.
MA X X, LI N, LIU S K, et al. Pyrrolic nitrogen-doped carbon sandwiched monolayer MoS2 vertically anchored on graphene oxide for high-performance sodium-ion battery anodes[J]. Journal of Solid State Electrochemistry, 2018, 22(9): 2801-2809.
ZHU Z Q, TANG Y X, LV Z S, et al. Fluoroethylene carbonate enabling a robust LiF-rich solid electrolyte interphase to enhance the stability of the MoS2 anode for lithium-ion storage[J]. Angewandte Chemie (International Ed in English), 2018, 57(14): 3656-3660.
WU J X, CIUCCI F, KIM J K. Molybdenum disulfide based nanomaterials for rechargeable batteries[J]. Chemistry (Weinheim an Der Bergstrasse, Germany), 2020, 26(29): 6296-6319.
XIE J R, ZHU K J, MIN J, et al. In-situ grown ultrathin MoS2 nanosheets on MoO2 hollow nanospheres to synthesize hierarchical nanostructures and its application in lithium-ion batteries[J]. Ionics, 2019, 25(4): 1487-1494.
LI Z Y, OTTMANN A, SUN Q, et al. Hierarchical MoS2-carbon porous nanorods towards atomic interfacial engineering for high-performance lithium storage[J]. Journal of Materials Chemistry A, 2019, 7(13): 7553-7564.
LONG F, CHEN Y, WU C H, et al. Unique three-dimensional hierarchical heterogeneous MoS2/graphene structures as a high-performance anode material for lithium-ion batteries[J]. Ionics, 2021, 27(5): 1977-1986.
CHOI M, HWANG J, SETIADI H, et al. One-pot synthesis of molybdenum disulfide-reduced graphene oxide (MoS2-RGO) composites and their high electrochemical performance as an anode in lithium ion batteries[J]. The Journal of Supercritical Fluids, 2017, 127: 81-89.
ZHAO Y, XU L, YAN J, et al. Facile preparation of NiFe2O4/MoS2 composite material with synergistic effect for high performance supercapacitor[J]. Journal of Alloys and Compounds, 2017, 726: 608-617.
QU B, SUN Y, LIU L L, et al. Ultrasmall Fe2O3 nanoparticles/MoS2 nanosheets composite as high-performance anode material for lithium ion batteries[J]. Scientific Reports, 2017, 7: doi: 10.1038/srep42772.
QIN S, LEI W W, LIU D, et al. Advanced N-doped mesoporous molybdenum disulfide nanosheets and the enhanced lithium-ion storage performance[J]. Journal of Materials Chemistry A, 2016, 4(4): 1440-1445.
WANG J C, ZHANG L Y, SUN K, et al. Improving ionic/electronic conductivity of MoS2 Li-ion anode via Manganese doping and structural optimization[J]. Chemical Engineering Journal, 2019, 372: 665-672.
XIA S S, WANG Y R, LIU Y, et al. Ultrathin MoS2 nanosheets tightly anchoring onto nitrogen-doped graphene for enhanced lithium storage properties[J]. Chemical Engineering Journal, 2018, 332: 431-439.
SHAO X J, WANG K D, PANG R, et al. Lithium intercalation in graphene/MoS2 composites: First-principles insights[J]. The Journal of Physical Chemistry C, 2015, 119(46): 25860-25867.
LIU Z X, GE D H, YANG P. Structure and interfacial properties investigation for ZnO/graphene interface[J]. Materials Chemistry and Physics, 2019, 229: 1-5.
ZHANG X E, ZHAO R F, WU Q H, et al. Petal-like MoS2 nanosheets space-confined in hollow mesoporous carbon spheres for enhanced lithium storage performance[J]. ACS Nano, 2017, 11(8): 8429-8436.
YOU Y, YE Y W, WEI M L, et al. Three-dimensional MoS2/rGO foams as efficient sulfur hosts for high-performance lithium-sulfur batteries[J]. Chemical Engineering Journal, 2019, 355: 671-678.
SUN Y M, HU X L, YU J C, et al. Morphosynthesis of a hierarchical MoO2 nanoarchitecture as a binder-free anode for lithium-ion batteries[J]. Energy & Environmental Science, 2011, 4(8): 2870-2877.
LIU B T, WANG S W, MO Q H, et al. Epitaxial MoS2 nanosheets on nitrogen doped graphite foam as a 3D electrode for highly efficient electrochemical hydrogen evolution[J]. Electrochimica Acta, 2018, 292: 407-418.
WAN Z M, SHAO J, YUN J J, et al. Core-shell structure of hierarchical quasi-hollow MoS2 microspheres encapsulated porous carbon as stable anode for Li-ion batteries[J]. Small (Weinheim an Der Bergstrasse, Germany), 2014, 10(23): 4975-4981.
WEI X, LIN C C, WU C W, et al. Three-dimensional hierarchically porous MoS2 foam as high-rate and stable lithium-ion battery anode[J]. Nature Communications, 2022, 13: doi: 10.1038/s41467-022-33790-z.
LIU Y C, ZHAO Y P, JIAO L F, et al. A graphene-like MoS2/graphene nanocomposite as a highperformance anode for lithium ion batteries[J]. Journal of Materials Chemistry A, 2014, 2(32): 13109-13115.
DING S J, CHEN JUN SONG, DAVID LOU X W. Glucose-assisted growth of MoS2 nanosheets on CNT backbone for improved lithium storage properties[J]. Chemistry-A European Journal, 2011, 17(47): 13142-13145.
CHAN K T, NEATON J B, COHEN M L. First-principles study of metal adatom adsorption on graphene[J]. Physical Review B, 2008, 77(23): doi: 10.1103/PhysRevB.77.235430.
PENG B, CHENG F Y, TAO Z L, et al. Lithium transport at silicon thin film: Barrier for high-rate capability anode[J]. The Journal of Chemical Physics, 2010, 133(3): doi: 10.1063/1.3462998.
SHAO Y F, GONG P L, PAN H, et al. H-/ dT-MoS2-on-MXene heterostructures as promising 2D anode materials for lithium-ion batteries: Insights from first principles[J]. Advanced Theory and Simulations, 2019, 2(8): doi: 10.1002/adts.201900045.
ZHANG X M, YU Z M, WANG S S, et al. Theoretical prediction of MoN2 monolayer as a high capacity electrode material for metal ion batteries[J]. Journal of Materials Chemistry A, 2016, 4(39): 15224-15231.
CUI Y H, ZHAO Y, CHEN H, et al. First-principles study of MoO3/graphene composite as cathode material for high-performance lithium-ion batteries[J]. Applied Surface Science, 2018, 433: 1083-1093.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}