Secondary batteries offer the advantages of high energy density and long cycle life, which have provided an effective solution for the storage and use of clean energy. To meet the ever-increasing demand of our society for energy, further in-depth research and development of high-energy-density secondary batteries are imminent. X-ray characterization techniques can provide a comprehensive insight into the research, design, and application of secondary batteries. This review summarizes relevant literature studies published in recent years, with a review of the latest progress and challenges of the X-ray spectroscopy methods in the research of secondary batteries. This review focuses on the technical principles, latest progress, and major scientific challenges of different X-ray characterization techniques, primarily including X-ray photoelectron spectroscopy, X-ray absorption spectroscopy, and resonant inelastic X-ray scattering. The technical characteristics, applicable conditions, and main advantages of different X-ray characterization methods are explained in detail, and the future application of X-ray spectroscopy in the field of secondary batteries is prospected. Comprehensive analysis shows that the X-ray technology provides various advanced characterization techniques that are sensitive and non-destructive to the lattice, electrons, and morphological structure of the electrode materials, and the structure of the electrode materials could be characterized from the macroscopic scale to the microscopic scale, reflecting the corresponding crystal structure and electronic structure evolution, charge compensation mechanism, ion and electron transport, and surface/interface chemical processes of electrode materials. Consequently, X-ray characterization techniques provide technical support for the design of high-performance secondary batteries.
CHEN Shuyuan. Research progress in the X-ray spectroscopy investigation of cathode materials for high-energy-density secondary batteries[J]. Energy Storage Science and Technology, 2024, 13(1): 113-129
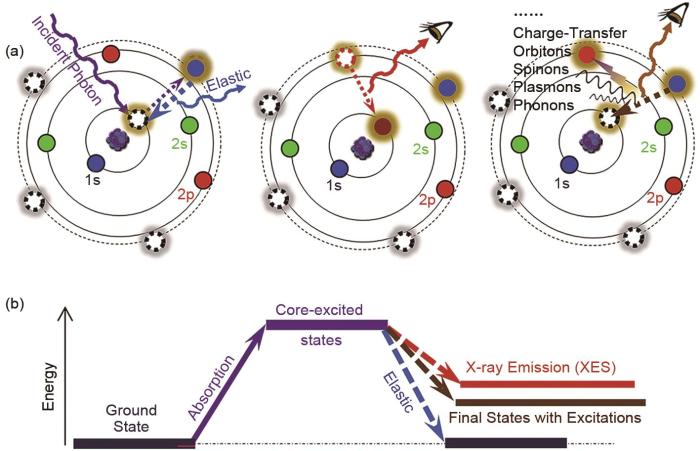
(a) photoelectric effect; (b) absorption of an X-ray photon results in excitation of a core electron into an unoccupied state; (c) Auger process; (d) fluorescent process[26]
XPS作为表面灵敏的分析技术,可以探测样品表面10 nm以内的信息。深度分析是材料分析中获取不同深度下组分和化学态信息的常用方法。在XPS深度分析中,有3个采样深度值得关注:①0~10 nm,这是基于常规Al Kα X射线XPS的探测深度,对于超薄膜层结构的深度分析可以通过变角度XPS实现;②0~30 nm,这是基于硬X射线XPS(HAXPES)的探测深度,可以通过切换X射线能量进行无损深度分析;③0~1000 nm,需要采用离子束剥离的破坏性深度剖析[29]。在SEI和CEI的深度分析中,可以采用不同能量的软硬X射线进行非破坏性的深度分析,当深度大于30 nm时,需要采用破坏性的深度剖析,常通过离子源(如Ar+/Xe+/Ar+GCIB)的轰击来移除表面原子进行破坏性深度分析。对于有机物组分的深度分析,单原子离子束会显著破坏共价键结构,需要采用Ar气团簇离子源(Ar-GCIB)或C60团簇离子源进行刻蚀。值得注意的是,当被溅射样品含有多种元素组分时,元素溅射差额的差异会导致择优溅射,导致样品的组成发生变化,例如电池材料中常见的氧化物物种CoO x 、NiO x 、MnO x 和FeO x 等在进行深度分析时容易发生择优溅射,导致Co/Ni/Mn/Fe等元素化学态被还原[30]。
Fig. 3
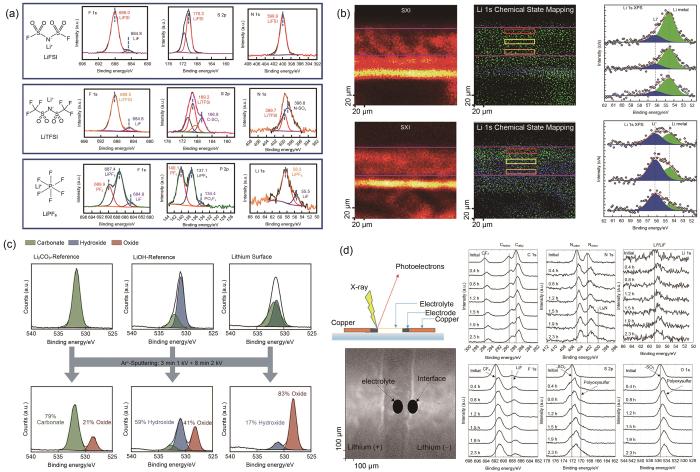
(a) X-ray photoelectron spectra of pristine salt films precipitated from liquid electrolytes (LiFSI、LiTFSI and LiPF6)[58]; (b) XPS chemical state mappings of Li 1s in the cross section of lithiated graphite anodes[61]; (c) comparison of the decomposition induced by argon sputtering for a Li2CO3-reference, a LiOH-reference and a lithium surface[62]; (d) C 1s, N 1s, Li 1s, F 1s, S 2p, and O 1s recorded at the interface Li/electrolyte during the polarization in the operando XPS cell[63]
锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应。例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物。对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行。Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池。该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试。XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力。在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响。
随着XPS空间分辨能力的提升,微区XPS技术可以获取样品表面的组分和化学态的局域化分布信息。Lu等[61]通过在石墨负极中构建基于Li x Cu6Sn5网络的固体锂传输通道大幅度降低了石墨负极的锂离子浓度梯度和相应的极化。利用微区XPS分析了电极截面上Li元素的化学态影像,金属Li0(54.66 eV)为主要化学态,而且沿截面均匀分布,显著降低了Li+浓度梯度和相应的极化效应[图3(b)]。
借助离子束剥离的XPS深度分析是研究SEI层组分和化学态在深度方向上分布的常用方法。Chen等[66]利用XPS深度分析研究了LiPF6-mixTHF电解质在负极上形成的LiF-有机双层SEI,研究结果表明SEI的顶部由有机物(RCH2OLi)和无机物(Li2O、LiF)两种组分组成,且碳含量(表示有机分解产物)随溅射时间增加而降低。但是,需要注意的是基于离子刻蚀的XPS深度分析是一个破坏过程,可能因为破坏化学态而导致产生假象。Yu等[58]指出Ar离子溅射可能改变SEI化学组成,导致锂盐的F 1s谱图中LiF峰的强度显著增加。类似的,Otto等[62]指出Ar离子溅射会导致LiOH和Li2CO3部分分解为Li2O[图3(c)]。此外,择优溅射效应会导致部分氧化物如CoO x 、NiO x 、MnO x 和FeO x 中的金属元素被还原。因此,深度分析中常用的Ar离子蚀刻的结果必须谨慎使用,因为离子溅射可能会引起副反应,导致高估表面上的LiF、Li2CO3和Li2O组分,以及对化学态的误判。Yin等[67]利用基于机械剥离方式的XPS深度分析证明了锂金属表面钙钛矿界面层的“钙钛矿-合金-锂金属”梯度渐变结构。由于离子刻蚀会导致ABX3钙钛矿B位离子的部分还原,这就导致基于Ar离子溅射的XPS深度分析无法真实客观地反映不同深度下B位元素的价态变化。因此,可以在手套箱惰性气氛的保护下利用Kapton胶带进行机械剥离,同时通过惰性气氛转移腔将制备好的样品从手套箱中转移到XPS实验装置中,逐层地暴露界面层在不同深度下的化学成分。结果表明表面处于氧化态(Sn2+和Pb2+),随着机械剥离深度增加,暴露出合金层(Sn0和Pb0)的比例逐渐增加。
图4
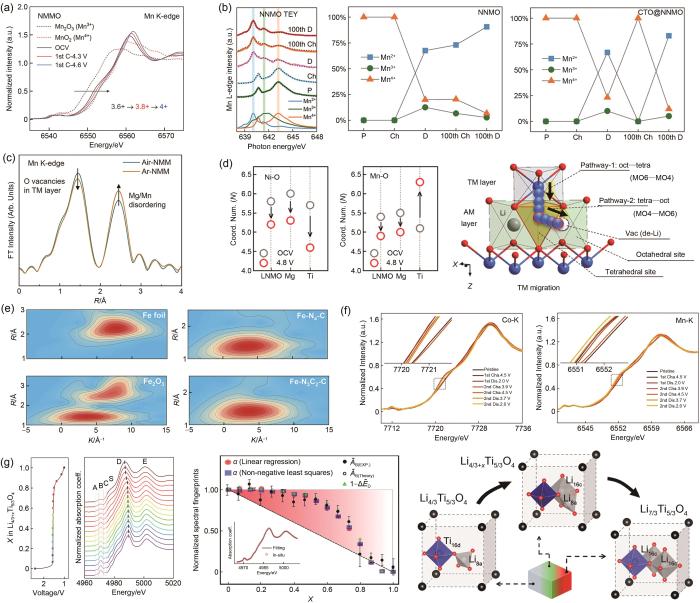
(a) NMMO在不同带电状态下的归一化Mn K边XANES光谱[71];(b) NNMO和CTO@NNMO在第1次和第100次充放电状态下的Mn L边XANES光谱和定量拟合结果[73];(c) Air-NMM和Ar-NMM的Mn K边傅里叶变换EXAFS谱[75];(d) 由EXAFS分析得出的Ni-O和Mn-O配位数的变化和TM的迁移途径[76];(e) Fe-N3C2-C、Fe-N4-C、Fe2O3 和Fe箔的WT图[77];(f) Na x LMNMT的Ni、Mn K边的原位XANES光谱[78];(g) 一系列Ti K边XANES,钛酸锂中Li嵌入过程中Li4/3Ti5/3O4 频谱加权函数随光谱指纹归一化幅度的演变以及锂化驱动的结构转变的演变[79]
Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states[71]; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle[73]; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM[75]; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration[76]; (e) WT plots of Fe- N3C2-C, Fe-N4-C, Fe2O3, and Fe foil[77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]
更重要的是,非原位状态表征难以清晰反映电池工况条件下的动态电子结构演变、离子及电子输运以及界面动态反应等关键信息,而原位XAS的成熟为探明电池充放电过程中真实的电荷补偿机制和复杂结构演变提供了可能。例如Guo等[78]利用Ni、Mn K边原位XANES以及EXAFS分析认为放电过程中P2-Na0.7Li0.03Mg0.03Ni0.27Mn0.6Ti0.07O2 (Na x LMNMT)中Mn未参与氧化还原反应,而Ni参与氧化还原反应并生成三价Ni进而导致J-T效应,使得晶体结构畸变[图4(f)]。Sun等[85]通过对比第1圈与第451圈xLi2MnO3·(1-x)LiMeO2(Me=Mn,Ni,Co) (LMR-NMC)充放电过程中Mn K边的原位XANES谱图,发现450圈的充放电循环后Mn的氧化还原活性被极大地削弱了,并通过对Co、Ni K边原位XANES的分析,认为450圈后的充放电过程中Co失去大量活性,并且Ni已完全失活,因此将450圈后LMR-NMC的容量归因于阴离子氧化还原。另外,Wang等[79]通过对Ti K边原位XANES中边前峰积分强度、主峰位置等参数的分析,发现Li4/3Ti5/3O4在嵌锂过程中并非发生简单的两相相变,而是在亚晶胞和颗粒尺度上发生多阶段的结构转变[图4(g)]。利用原位XAS分析能深入理解钠离子电池电极材料的电荷补偿机制与容量衰减机制,优化电池材料组成与结构,为进一步提升电池性能提供重要科学依据。
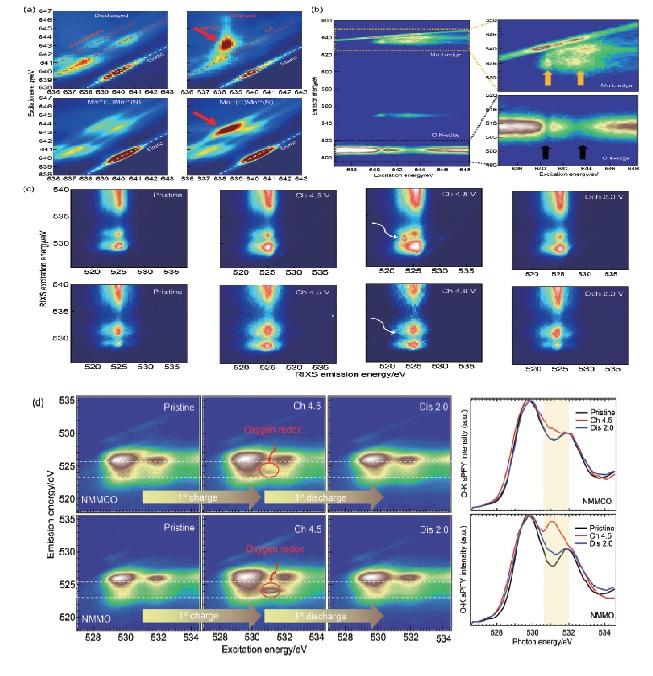
RIXS技术能对不同激发能的发射光子进行分辨和统计,并进一步获得更高维度的信息,从而有效解决sXAS难以分辨的价态信息。例如,Wessells等[86]通过Mn L3边mRIXS谱图对Na x Mn[Mn(CN)6]0.81(MnHCMn)正极材料中Mn在充放电过程中的价态进行表征,发现放电过程中mRIXS谱图中出现多组平行于弹性峰的d-d激发特征区域,说明t2g和eg态被部分占据,证明了二价Mn的存在;充电过程中,mRIXS谱图在激发能为643.5 eV时具有较高的能量损失,这种特征对应于Mn1+低自旋3d6体系的d-d激发,反映该体系的t2g态被完全占据,从而证明了MnHCMn中一价Mn的存在[图5(a)]。类似地,Yang等[49]通过对比发现,sXAS技术并不能区分一价Mn与二价Mn,这说明RIXS技术在价态分辨方面有不可替代的作用。
Fig. 5
(a) Mn L3-edge RIXS maps and corresponding RIXS calculations on MnHCMn electrodes at the discharged and charged states[86]; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge[87]; (c) O K-edge RIXS maps of a Li1.2Ni0.2Mn0.6O2 and Li1.2Ni0.2Ru0.6O2 electrodes at different states of charge[88]; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge[84]
CEN G J, QIAO R H, SHEN X Y, et al. Reviews of selected 100 recent papers for lithium batteries(Jun. 1, 2023 to Jul. 31, 2023)[J]. Energy Storage Science and Technology, 2023, 12(9): 3003-3018.
YU Z J, WANG W, ZHU Y, et al. Construction of double reaction zones for long-life quasi-solid aluminum-ion batteries by realizing maximum electron transfer[J]. Nature Communications, 2023, 14: 5596.
ZHENG W, LIU Q, LU Z G. Modulating anionic redox reaction in layered transition metal oxides for sodium-ion batteries[J]. Energy Storage Science and Technology, 2020, 9(5): 1416-1427.
MA X, YUAN C, LIU G L, et al. Steering the liquid-solid redox conversion of lithium-selenium batteries through ultrafine MoC catalyst[J]. Chemical Communications, 2023, 59: 11208-11.
YAN T R, FENG J, ZENG P, et al. Modulating eg orbitals through ligand engineering to boost the electrocatalytic activity of NiSe for advanced lithium-sulfur batteries[J]. Journal of Energy Chemistry, 2022, 74: 317-323.
YAN T R, WU Y, GONG F, et al. TiH2 nanodots exfoliated via facile sonication as bifunctional electrocatalysts for Li-S batteries[J]. ACS Applied Materials & Interfaces, 2022, 14(5): 6937-6944.
ZHU X H, ZHUANG Y H, ZHAO Y, et al. Development of layered cathode materials for sodium-ion batteries[J]. Energy Storage Science and Technology, 2020, 9(5): 1340-1349.
FUJITA T, TODA K. Microdisplacement measurement using a liquid-delay-line oscillator[J]. Japanese Journal of Applied Physics, 2003, 42(Part 1, No. 9B): 6131-6134.
YANG S C, HE R, ZHANG Z J, et al. CHAIN: Cyber hierarchy and interactional network enabling digital solution for battery full-lifespan management[J]. Matter, 2020, 3(1): 27-41.
WHITTINGHAM M S. Lithium batteries and cathode materials[J]. Chemical Reviews, 2004, 104(10): 4271-4302.
ZENG P, SU B, WANG X L, et al. In situ reconstruction of electrocatalysts for lithium-sulfur batteries: Progress and prospects[J]. Advanced Functional Materials, 2023, 33(33): 2301743.
XIANG X D, ZHANG K, CHEN J. Recent advances and prospects of cathode materials for sodium-ion batteries[J]. Advanced Materials (Deerfield Beach, Fla), 2015, 27(36): 5343-5364.
KIM S W, SEO D H, MA X H, et al. Electrode materials for rechargeable sodium-ion batteries: Potential alternatives to current lithium-ion batteries[J]. Advanced Energy Materials, 2012, 2(7): 710-721.
FANG K, TANG Y L, LIU J J, et al. Injecting excess Na into a P2-type layered oxide cathode to achieve presodiation in a Na-ion full cell[J]. Nano Letters, 2023, 23(14): 6681-6688.
YUAN C, SONG X C, ZENG P, et al. Precisely optimizing polysulfides adsorption and conversion by local coordination engineering for high-performance Li-S batteries[J]. Nano Energy, 2023, 110: 108353.
DING L Y, WANG L, GAO J C, et al. Facile Zn2+ desolvation enabled by local coordination engineering for long-cycling aqueous zinc-ion batteries[J]. Advanced Functional Materials, 2023, 33(32): 2301648.
BRUNDLE C R, CHUANG T J, RICE D W. X- ray photoemission study of the interaction of oxygen and air with clean cobalt surfaces[J]. Surface Science, 1976, 60(2): 286-300.
LIN F, LIU Y J, YU X Q, et al. Synchrotron X-ray analytical techniques for studying materials electrochemistry in rechargeable batteries[J]. Chemical Reviews, 2017, 117(21): 13123-13186.
SIOL S, MANN J, NEWMAN J, et al. Concepts for chemical state analysis at constant probing depth by lab-based XPS/HAXPES combining soft and hard X-ray sources[J]. Surface and Interface Analysis, 2020, 52(12): 802-810.
TIMOSHENKO J, ROLDAN CUENYA B. in situ/Operando electrocatalyst characterization by X-ray absorption spectroscopy[J]. Chemical Reviews, 2021, 121(2): 882-961.
SONG Z X, LI J J, DAVIS K D, et al. Emerging applications of synchrotron radiation X-ray techniques in single atomic catalysts[J]. Small Methods, 2022, 6(11): e2201078.
GIORGETTI M. A review on the structural studies of batteries and host materials by X-ray absorption spectroscopy[J]. ISRN Materials Science, 2013, 2013: 1-22.
HARKS P P R M L, MULDER F M, NOTTEN P H L. in situ methods for Li-ion battery research: A review of recent developments[J]. Journal of Power Sources, 2015, 288: 92-105.
GONG Z L, YANG Y. The application of synchrotron X-ray techniques to the study of rechargeable batteries[J]. Journal of Energy Chemistry, 2018, 27(6): 1566-1583.
ZHANG L, GUO J H. Understanding the reaction mechanism of lithium-sulfur batteries by in situ/operando X-ray absorption spectroscopy[J]. Arabian Journal for Science and Engineering, 2019, 44(7): 6217-6229.
YANG W L, DEVEREAUX T P. Anionic and cationic redox and interfaces in batteries: Advances from soft X-ray absorption spectroscopy to resonant inelastic scattering[J]. Journal of Power Sources, 2018, 389: 188-197.
BAKER M L, MARA M W, YAN J J, et al. K- and L-edge X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) determination of differential orbital covalency (DOC) of transition metal sites[J]. Coordination Chemistry Reviews, 2017, 345: 182-208.
BENKERT A, MEYER F, HAUSCHILD D, et al. Isotope effects in the resonant inelastic soft X-ray scattering maps of gas-phase methanol[J]. The Journal of Physical Chemistry A, 2016, 120(14): 2260-2267.
LIU Y S, GLANS P A, CHUANG C H, et al. Perspectives of in situ/operando resonant inelastic X-ray scattering in catalytic energy materials science[J]. Journal of Electron Spectroscopy and Related Phenomena, 2015, 200: 282-292.
KUNNUS K, ZHANG W K, DELCEY M G, et al. Viewing the valence electronic structure of ferric and ferrous hexacyanide in solution from the Fe and cyanide perspectives[J]. The Journal of Physical Chemistry B, 2016, 120(29): 7182-7194.
MATSUBARA M, UOZUMI T, KOTANI A, et al. Polarization dependence of resonant X-ray emission spectra in 3dn transition metal compounds with n = 0, 1, 2, 3[J]. Journal of the Physical Society of Japan, 2002, 71(1): 347-356.
GHIRINGHELLI G, MATSUBARA M, DALLERA C, et al. Resonant inelastic X-ray scattering of MnO: L2, 3 edge measurements and assessment of their interpretation[J]. Physical Review B, 2006, 73(3): 035111.
KUNNUS K, JOSEFSSON I, SCHRECK S, et al. From ligand fields to molecular orbitals: Probing the local valence electronic structure of Ni2+ in aqueous solution with resonant inelastic X-ray scattering[J]. The Journal of Physical Chemistry B, 2013, 117(51): 16512-16521.
GLATZEL P, SINGH J, KVASHNINA K O, et al. In situ characterization of the 5d density of states of Pt nanoparticles upon adsorption of CO[J]. Journal of the American Chemical Society, 2010, 132(8): 2555-2557.
KVASHNINA K O, BUTORIN S M, GLATZEL P. Direct study of the f-electron configuration in lanthanide systems[J]. Journal of Analytical Atomic Spectrometry, 2011, 26(6): 1265-1272.
WU J P, SHEN Z X, YANG W L. Redox mechanism in Na-ion battery cathodes probed by advanced soft X-ray spectroscopy[J]. Frontiers in Chemistry, 2020, 8: 816.
AMENT L J P, VAN VEENENDAAL M, DEVEREAUX T P, et al. Resonant inelastic X-ray scattering studies of elementary excitations[J]. Reviews of Modern Physics, 2011, 83(2): 705-767.
DE GROOT F. High-resolution X-ray emission and X-ray absorption spectroscopy[J]. Chemical Reviews, 2001, 101(6): 1779-1808.
ÅGREN H, LUO Y, GELMUKHANOV F, et al. Screening in resonant X-ray emission of molecules[J]. Journal of Electron Spectroscopy and Related Phenomena, 1996, 82(1/2): 125-134.
FÖHLISCH A, HASSELSTRÖM J, BENNICH P, et al. Ground-state interpretation of X-ray emission spectroscopy on adsorbates: CO adsorbed on Cu(100)[J]. Physical Review B, 2000, 61(23): 16229-16240.
SHUTTHANANDAN V, NANDASIRI M, ZHENG J M, et al. Applications of XPS in the characterization of battery materials[J]. Journal of Electron Spectroscopy and Related Phenomena, 2019, 231: 2-10.
ATKINS D, AYERBE E, BENAYAD A, et al. Understanding battery interfaces by combined characterization and simulation approaches: Challenges and perspectives[J]. Advanced Energy Materials, 2022, 12(17): 2102687.
WOOD K N, TEETER G. XPS on Li-battery-related compounds: Analysis of inorganic SEI phases and a methodology for charge correction[J]. ACS Applied Energy Materials, 2018, 1(9): 4493-4504.
YU W L, YU Z A, CUI Y, et al. Degradation and speciation of Li salts during XPS analysis for battery research[J]. ACS Energy Letters, 2022, 7(10): 3270-3275.
HAN J G, BIN LEE J, CHA A M, et al. Unsymmetrical fluorinated malonatoborate as an amphoteric additive for high-energy-density lithium-ion batteries[J]. Energy & Environmental Science, 2018, 11(6): 1552-1562.
ZHENG Y, HE Y B, QIAN K, et al. Effects of state of charge on the degradation of LiFePO4/graphite batteries during accelerated storage test[J]. Journal of Alloys and Compounds, 2015, 639: 406-414.
LU L L, ZHU Z X, MA T, et al. Superior fast-charging lithium-ion batteries enabled by the high-speed solid-state lithium transport of an intermetallic Cu6Sn5 network[J]. Advanced Materials, 2022, 34(32): 2202688.
OTTO S K, MORYSON Y, KRAUSKOPF T, et al. In-depth characterization of lithium-metal surfaces with XPS and ToF-SIMS: Toward better understanding of the passivation layer[J]. Chemistry of Materials, 2021, 33(3): 859-867.
BENAYAD A, MORALES-UGARTE J E, SANTINI C C, et al. Operando XPS: A novel approach for probing the lithium/electrolyte interphase dynamic evolution[J]. The Journal of Physical Chemistry A, 2021, 125(4): 1069-1081.
CHO D H, JO C H, CHO W, et al. Effect of residual lithium compounds on layer Ni-rich Li[Ni0.7Mn0.3]O2[J]. Journal of the Electrochemical Society, 2014, 161(6): A920-A926.
CHEN J, FAN X L, LI Q, et al. Electrolyte design for LiF-rich solid-electrolyte interfaces to enable high-performance microsized alloy anodes for batteries[J]. Nature Energy, 2020, 5(5): 386-397.
YIN Y C, WANG Q, YANG J T, et al. Metal chloride perovskite thin film based interfacial layer for shielding lithium metal from liquid electrolyte[J]. Nature Communications, 2020, 11: 1761.
MALMGREN S, CIOSEK K, HAHLIN M, et al. Comparing anode and cathode electrode/electrolyte interface composition and morphology using soft and hard X-ray photoelectron spectroscopy[J]. Electrochimica Acta, 2013, 97: 23-32.
QIAN Y X, CHU Y L, ZHENG Z T, et al. A new cyclic carbonate enables high power/low temperature lithium-ion batteries[J]. Energy Storage Materials, 2022, 45: 14-23.
IIDA S I, TERASHIMA M, MAMIYA K, et al. Characterization of cathode-electrolyte interface in all-solid-state batteries using TOF-SIMS, XPS, and UPS/LEIPS[J]. Journal of Vacuum Science & Technology B, 2021, 39(4): doi: 10.1116/6.0001044.
WU Z H, NI Y X, TAN S, et al. Realizing high capacity and zero strain in layered oxide cathodes via lithium dual-site substitution for sodium-ion batteries[J]. Journal of the American Chemical Society, 2023, 145(17): 9596-9606.
ZOU P C, YAO L B, WANG C Y, et al. Regulating cation interactions for zero-strain and high-voltage P2-type Na2/3Li1/6Co1/6Mn2/3O2 layered oxide cathodes of sodium-ion batteries[J]. Angewandte Chemie International Edition, 2023, 62(28): e202304628.
HU H L, HE H C, XIE R K, et al. Achieving reversible Mn2+/Mn4+ double redox couple through anionic substitution in a P2-type layered oxide cathode[J]. Nano Energy, 2022, 99: 107390.
JIN J T, LIU Y C, ZHAO X D, et al. Annealing in argon universally upgrades the Na-storage performance of Mn-based layered oxide cathodes by creating bulk oxygen vacancies[J]. Angewandte Chemie (International Ed in English), 2023, 62(15): e202219230.
ZHANG B D, ZHANG Y M, WANG X T, et al. Role of substitution elements in enhancing the structural stability of Li-rich layered cathodes[J]. Journal of the American Chemical Society, 2023: 8700-8713.
LIU G L, WANG W M, ZENG P, et al. Strengthened d–p orbital hybridization through asymmetric coordination engineering of single-atom catalysts for durable lithium-sulfur batteries[J]. Nano Letters, 2022, 22(15): 6366-6374.
CHENG Z W, ZHAO B, GUO Y J, et al. Mitigating the large-volume phase transition of P2-type cathodes by synergetic effect of multiple ions for improved sodium-ion batteries[J]. Advanced Energy Materials, 2022, 12(14): 2103461.
ZHANG W, TOPSAKAL M, CAMA C, et al. Multi-stage structural transformations in zero-strain lithium titanate unveiled by in situ X-ray absorption fingerprints[J]. Journal of the American Chemical Society, 2017, 139(46): 16591-16603.
HU H L, KAO C W, CHENG C, et al. Local construction of Mn-based layered cathodes through covalency modulation for sodium-ion batteries[J]. ACS Applied Materials & Interfaces, 2023, 15(25): 30332-30341.
XU C L, HUA W B, ZHANG Q H, et al. Sufficient utilization of Mn2+/Mn3+/Mn4+ redox in NASICON phosphate cathodes towards high-energy Na-ions batteries[J]. Advanced Functional Materials, 2023, 33(33): 2302810.
ZHAO G, CHEN Q J, WANG L, et al. Self-standing sulfur cathodes enabled by a single Fe site decorated fibrous membrane for durable lithium-sulfur batteries[J]. Journal of Materials Chemistry A, 2022, 10(37): 19893-19902.
DU Z Z, CHEN X J, HU W, et al. Cobalt in nitrogen-doped graphene as single-atom catalyst for high-sulfur content lithium–sulfur batteries[J]. Journal of the American Chemical Society, 2019, 141(9): 3977-3985.
CHENG C, CHEN C, CHU S Y, et al. Enhancing the reversibility of lattice oxygen redox through modulated transition metal-oxygen covalency for layered battery electrodes[J]. Advanced Materials, 2022, 34(20): e2201152.
XIAO B W, LIU H S, CHEN N, et al. Size-mediated recurring spinel sub-nanodomains in Li- and Mn-rich layered cathode materials[J]. Angewandte Chemie International Edition, 2020, 59(34): 14313-14320.
FIROUZI A, QIAO R M, MOTALLEBI S, et al. Monovalent manganese based anodes and co-solvent electrolyte for stable low-cost high-rate sodium-ion batteries[J]. Nature Communications, 2018, 9: 861.
CHENG C, LI S Y, LIU T F, et al. Elucidation of anionic and cationic redox reactions in a prototype sodium-layered oxide cathode[J]. ACS Applied Materials & Interfaces, 2019, 11(44): 41304-41312.
DAI K H, SHAO W W, ZHAO B B, et al. Precisely quantifying bulk transition metal valence evolution in conventional battery electrode by inverse partial fluorescence yield[J]. Journal of Energy Chemistry, 2022, 69: 363-368.
JI H W, WU J P, CAI Z J, et al. Ultrahigh power and energy density in partially ordered lithium-ion cathode materials[J]. Nature Energy, 2020, 5(3): 213-221.
WU J P, ZHUO Z Q, RONG X H, et al. Dissociate lattice oxygen redox reactions from capacity and voltage drops of battery electrodes[J]. Science Advances, 2020, 6(6): eaaw3871.
DAI K H, MAO J, ZHUO Z Q, et al. Negligible voltage hysteresis with strong anionic redox in conventional battery electrode[J]. Nano Energy, 2020, 74: 104831.
DAI K H, WU J P, ZHUO Z Q, et al. High reversibility of lattice oxygen redox quantified by direct bulk probes of both anionic and cationic redox reactions[J]. Joule, 2019, 3(2): 518-541.
CHEN M Z, CHOU S L, DOU S X. Understanding challenges of cathode materials for sodium-ion batteries using synchrotron-based X-ray absorption spectroscopy[J]. Batteries & Supercaps, 2019, 2(10): 842-851.
GHIGNA P, QUARTARONE E. Operando X-ray absorption spectroscopy on battery materials: A review of recent developments[J]. Journal of Physics: Energy, 2021, 3(3): 032006.
... (a) photoelectric effect; (b) absorption of an X-ray photon results in excitation of a core electron into an unoccupied state; (c) Auger process; (d) fluorescent process[26] ...
... XPS作为表面灵敏的分析技术,可以探测样品表面10 nm以内的信息.深度分析是材料分析中获取不同深度下组分和化学态信息的常用方法.在XPS深度分析中,有3个采样深度值得关注:①0~10 nm,这是基于常规Al Kα X射线XPS的探测深度,对于超薄膜层结构的深度分析可以通过变角度XPS实现;②0~30 nm,这是基于硬X射线XPS(HAXPES)的探测深度,可以通过切换X射线能量进行无损深度分析;③0~1000 nm,需要采用离子束剥离的破坏性深度剖析[29].在SEI和CEI的深度分析中,可以采用不同能量的软硬X射线进行非破坏性的深度分析,当深度大于30 nm时,需要采用破坏性的深度剖析,常通过离子源(如Ar+/Xe+/Ar+GCIB)的轰击来移除表面原子进行破坏性深度分析.对于有机物组分的深度分析,单原子离子束会显著破坏共价键结构,需要采用Ar气团簇离子源(Ar-GCIB)或C60团簇离子源进行刻蚀.值得注意的是,当被溅射样品含有多种元素组分时,元素溅射差额的差异会导致择优溅射,导致样品的组成发生变化,例如电池材料中常见的氧化物物种CoO x 、NiO x 、MnO x 和FeO x 等在进行深度分析时容易发生择优溅射,导致Co/Ni/Mn/Fe等元素化学态被还原[30]. ...
1
... XPS作为表面灵敏的分析技术,可以探测样品表面10 nm以内的信息.深度分析是材料分析中获取不同深度下组分和化学态信息的常用方法.在XPS深度分析中,有3个采样深度值得关注:①0~10 nm,这是基于常规Al Kα X射线XPS的探测深度,对于超薄膜层结构的深度分析可以通过变角度XPS实现;②0~30 nm,这是基于硬X射线XPS(HAXPES)的探测深度,可以通过切换X射线能量进行无损深度分析;③0~1000 nm,需要采用离子束剥离的破坏性深度剖析[29].在SEI和CEI的深度分析中,可以采用不同能量的软硬X射线进行非破坏性的深度分析,当深度大于30 nm时,需要采用破坏性的深度剖析,常通过离子源(如Ar+/Xe+/Ar+GCIB)的轰击来移除表面原子进行破坏性深度分析.对于有机物组分的深度分析,单原子离子束会显著破坏共价键结构,需要采用Ar气团簇离子源(Ar-GCIB)或C60团簇离子源进行刻蚀.值得注意的是,当被溅射样品含有多种元素组分时,元素溅射差额的差异会导致择优溅射,导致样品的组成发生变化,例如电池材料中常见的氧化物物种CoO x 、NiO x 、MnO x 和FeO x 等在进行深度分析时容易发生择优溅射,导致Co/Ni/Mn/Fe等元素化学态被还原[30]. ...
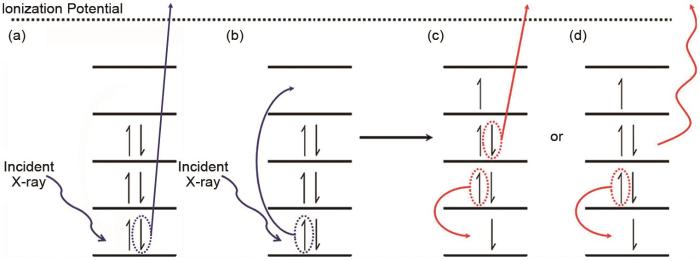
... [37](a) Atomic models of RIXS with different deactivation modes; (b) different states involved in the RIXS process<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 2
... (c) Ar离子溅射导致LiOH、Li2CO3 和锂片成分分解对比结果[62];(d) 原位XPS电池在极化条件下锂/电解质界面处C 1s、N 1s、Li 1s、F 1s、S 2p和O 1s谱图[63]<strong>(a) X-ray photoelectron spectra of pristine salt films precipitated from liquid electrolytes (LiFSI</strong>、<strong>LiTFSI and LiPF<sub>6</sub>)</strong><sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup><strong>; (b) XPS chemical state mappings of Li 1s in the cross section of lithiated graphite anodes</strong><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup><strong>; (c) comparison of the decomposition induced by argon sputtering for a Li<sub>2</sub>CO<sub>3</sub>-reference, a LiOH-reference and a lithium surface</strong><sup>[<xref ref-type="bibr" rid="R62">62</xref>]</sup><strong>; (d) C 1s, N 1s, Li 1s, F 1s, S 2p, and O 1s recorded at the interface Li/electrolyte during the polarization in the operando XPS cell</strong><sup>[<xref ref-type="bibr" rid="R63">63</xref>]</sup>Fig. 3
锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
... 借助离子束剥离的XPS深度分析是研究SEI层组分和化学态在深度方向上分布的常用方法.Chen等[66]利用XPS深度分析研究了LiPF6-mixTHF电解质在负极上形成的LiF-有机双层SEI,研究结果表明SEI的顶部由有机物(RCH2OLi)和无机物(Li2O、LiF)两种组分组成,且碳含量(表示有机分解产物)随溅射时间增加而降低.但是,需要注意的是基于离子刻蚀的XPS深度分析是一个破坏过程,可能因为破坏化学态而导致产生假象.Yu等[58]指出Ar离子溅射可能改变SEI化学组成,导致锂盐的F 1s谱图中LiF峰的强度显著增加.类似的,Otto等[62]指出Ar离子溅射会导致LiOH和Li2CO3部分分解为Li2O[图3(c)].此外,择优溅射效应会导致部分氧化物如CoO x 、NiO x 、MnO x 和FeO x 中的金属元素被还原.因此,深度分析中常用的Ar离子蚀刻的结果必须谨慎使用,因为离子溅射可能会引起副反应,导致高估表面上的LiF、Li2CO3和Li2O组分,以及对化学态的误判.Yin等[67]利用基于机械剥离方式的XPS深度分析证明了锂金属表面钙钛矿界面层的“钙钛矿-合金-锂金属”梯度渐变结构.由于离子刻蚀会导致ABX3钙钛矿B位离子的部分还原,这就导致基于Ar离子溅射的XPS深度分析无法真实客观地反映不同深度下B位元素的价态变化.因此,可以在手套箱惰性气氛的保护下利用Kapton胶带进行机械剥离,同时通过惰性气氛转移腔将制备好的样品从手套箱中转移到XPS实验装置中,逐层地暴露界面层在不同深度下的化学成分.结果表明表面处于氧化态(Sn2+和Pb2+),随着机械剥离深度增加,暴露出合金层(Sn0和Pb0)的比例逐渐增加. ...
... (c) Ar离子溅射导致LiOH、Li2CO3 和锂片成分分解对比结果[62];(d) 原位XPS电池在极化条件下锂/电解质界面处C 1s、N 1s、Li 1s、F 1s、S 2p和O 1s谱图[63]<strong>(a) X-ray photoelectron spectra of pristine salt films precipitated from liquid electrolytes (LiFSI</strong>、<strong>LiTFSI and LiPF<sub>6</sub>)</strong><sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup><strong>; (b) XPS chemical state mappings of Li 1s in the cross section of lithiated graphite anodes</strong><sup>[<xref ref-type="bibr" rid="R61">61</xref>]</sup><strong>; (c) comparison of the decomposition induced by argon sputtering for a Li<sub>2</sub>CO<sub>3</sub>-reference, a LiOH-reference and a lithium surface</strong><sup>[<xref ref-type="bibr" rid="R62">62</xref>]</sup><strong>; (d) C 1s, N 1s, Li 1s, F 1s, S 2p, and O 1s recorded at the interface Li/electrolyte during the polarization in the operando XPS cell</strong><sup>[<xref ref-type="bibr" rid="R63">63</xref>]</sup>Fig. 3
锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
... 随着XPS空间分辨能力的提升,微区XPS技术可以获取样品表面的组分和化学态的局域化分布信息.Lu等[61]通过在石墨负极中构建基于Li x Cu6Sn5网络的固体锂传输通道大幅度降低了石墨负极的锂离子浓度梯度和相应的极化.利用微区XPS分析了电极截面上Li元素的化学态影像,金属Li0(54.66 eV)为主要化学态,而且沿截面均匀分布,显著降低了Li+浓度梯度和相应的极化效应[图3(b)]. ...
... [62]; (d) C 1s, N 1s, Li 1s, F 1s, S 2p, and O 1s recorded at the interface Li/electrolyte during the polarization in the operando XPS cell[63]Fig. 3
锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
... 借助离子束剥离的XPS深度分析是研究SEI层组分和化学态在深度方向上分布的常用方法.Chen等[66]利用XPS深度分析研究了LiPF6-mixTHF电解质在负极上形成的LiF-有机双层SEI,研究结果表明SEI的顶部由有机物(RCH2OLi)和无机物(Li2O、LiF)两种组分组成,且碳含量(表示有机分解产物)随溅射时间增加而降低.但是,需要注意的是基于离子刻蚀的XPS深度分析是一个破坏过程,可能因为破坏化学态而导致产生假象.Yu等[58]指出Ar离子溅射可能改变SEI化学组成,导致锂盐的F 1s谱图中LiF峰的强度显著增加.类似的,Otto等[62]指出Ar离子溅射会导致LiOH和Li2CO3部分分解为Li2O[图3(c)].此外,择优溅射效应会导致部分氧化物如CoO x 、NiO x 、MnO x 和FeO x 中的金属元素被还原.因此,深度分析中常用的Ar离子蚀刻的结果必须谨慎使用,因为离子溅射可能会引起副反应,导致高估表面上的LiF、Li2CO3和Li2O组分,以及对化学态的误判.Yin等[67]利用基于机械剥离方式的XPS深度分析证明了锂金属表面钙钛矿界面层的“钙钛矿-合金-锂金属”梯度渐变结构.由于离子刻蚀会导致ABX3钙钛矿B位离子的部分还原,这就导致基于Ar离子溅射的XPS深度分析无法真实客观地反映不同深度下B位元素的价态变化.因此,可以在手套箱惰性气氛的保护下利用Kapton胶带进行机械剥离,同时通过惰性气氛转移腔将制备好的样品从手套箱中转移到XPS实验装置中,逐层地暴露界面层在不同深度下的化学成分.结果表明表面处于氧化态(Sn2+和Pb2+),随着机械剥离深度增加,暴露出合金层(Sn0和Pb0)的比例逐渐增加. ...
锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
... 锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
1
... 锂离子电池中部分材料具有高反应活性,容易与空气中的水和氧气等发生反应.例如,Cho等[64]利用XPS研究了高镍三元正极材料,发现如果暴露在空气中,材料表面的残锂会与空气中的CO2、H2O发生反应形成LiOH、Li2CO3等副产物.对于大气敏感样品,可靠和可重复的表征是具有挑战性的,因为样品的制备、转移和分析必须在不与空气气氛接触的情况下进行.Yin等[65]设计开发出镧系金属卤化物基固态电解质新家族Li x M y Ln z Cl3,可直接与锂金属负极和三元正极匹配,实现无任何电极修饰且室温可运行的全固态锂金属电池.该工作利用XPS研究了Li/SEI界面的稳定性机制,并利用惰性气氛转移管将样品进行样品转移,同时为验证转移管的密封能力的可靠性,在样品托上附加了一块新鲜的金属钠片作为参照样品,随样品一起由手套箱转移到XPS设备进行测试.XPS结果表明钠参照样品中金属Na比例高达95.2%,很好地证明了样品转移管的密封能力.在Li/SEI的表面检测到86.6%的Ta5+,表明界面钝化层可以有效地缓解锂沉积/剥离过程中的界面应变,并保护SEI免受锂金属的影响. ...
1
... 借助离子束剥离的XPS深度分析是研究SEI层组分和化学态在深度方向上分布的常用方法.Chen等[66]利用XPS深度分析研究了LiPF6-mixTHF电解质在负极上形成的LiF-有机双层SEI,研究结果表明SEI的顶部由有机物(RCH2OLi)和无机物(Li2O、LiF)两种组分组成,且碳含量(表示有机分解产物)随溅射时间增加而降低.但是,需要注意的是基于离子刻蚀的XPS深度分析是一个破坏过程,可能因为破坏化学态而导致产生假象.Yu等[58]指出Ar离子溅射可能改变SEI化学组成,导致锂盐的F 1s谱图中LiF峰的强度显著增加.类似的,Otto等[62]指出Ar离子溅射会导致LiOH和Li2CO3部分分解为Li2O[图3(c)].此外,择优溅射效应会导致部分氧化物如CoO x 、NiO x 、MnO x 和FeO x 中的金属元素被还原.因此,深度分析中常用的Ar离子蚀刻的结果必须谨慎使用,因为离子溅射可能会引起副反应,导致高估表面上的LiF、Li2CO3和Li2O组分,以及对化学态的误判.Yin等[67]利用基于机械剥离方式的XPS深度分析证明了锂金属表面钙钛矿界面层的“钙钛矿-合金-锂金属”梯度渐变结构.由于离子刻蚀会导致ABX3钙钛矿B位离子的部分还原,这就导致基于Ar离子溅射的XPS深度分析无法真实客观地反映不同深度下B位元素的价态变化.因此,可以在手套箱惰性气氛的保护下利用Kapton胶带进行机械剥离,同时通过惰性气氛转移腔将制备好的样品从手套箱中转移到XPS实验装置中,逐层地暴露界面层在不同深度下的化学成分.结果表明表面处于氧化态(Sn2+和Pb2+),随着机械剥离深度增加,暴露出合金层(Sn0和Pb0)的比例逐渐增加. ...
1
... 借助离子束剥离的XPS深度分析是研究SEI层组分和化学态在深度方向上分布的常用方法.Chen等[66]利用XPS深度分析研究了LiPF6-mixTHF电解质在负极上形成的LiF-有机双层SEI,研究结果表明SEI的顶部由有机物(RCH2OLi)和无机物(Li2O、LiF)两种组分组成,且碳含量(表示有机分解产物)随溅射时间增加而降低.但是,需要注意的是基于离子刻蚀的XPS深度分析是一个破坏过程,可能因为破坏化学态而导致产生假象.Yu等[58]指出Ar离子溅射可能改变SEI化学组成,导致锂盐的F 1s谱图中LiF峰的强度显著增加.类似的,Otto等[62]指出Ar离子溅射会导致LiOH和Li2CO3部分分解为Li2O[图3(c)].此外,择优溅射效应会导致部分氧化物如CoO x 、NiO x 、MnO x 和FeO x 中的金属元素被还原.因此,深度分析中常用的Ar离子蚀刻的结果必须谨慎使用,因为离子溅射可能会引起副反应,导致高估表面上的LiF、Li2CO3和Li2O组分,以及对化学态的误判.Yin等[67]利用基于机械剥离方式的XPS深度分析证明了锂金属表面钙钛矿界面层的“钙钛矿-合金-锂金属”梯度渐变结构.由于离子刻蚀会导致ABX3钙钛矿B位离子的部分还原,这就导致基于Ar离子溅射的XPS深度分析无法真实客观地反映不同深度下B位元素的价态变化.因此,可以在手套箱惰性气氛的保护下利用Kapton胶带进行机械剥离,同时通过惰性气氛转移腔将制备好的样品从手套箱中转移到XPS实验装置中,逐层地暴露界面层在不同深度下的化学成分.结果表明表面处于氧化态(Sn2+和Pb2+),随着机械剥离深度增加,暴露出合金层(Sn0和Pb0)的比例逐渐增加. ...
... [71];(b) NNMO和CTO@NNMO在第1次和第100次充放电状态下的Mn L边XANES光谱和定量拟合结果[73];(c) Air-NMM和Ar-NMM的Mn K边傅里叶变换EXAFS谱[75];(d) 由EXAFS分析得出的Ni-O和Mn-O配位数的变化和TM的迁移途径[76];(e) Fe-N3C2-C、Fe-N4-C、Fe2O3 和Fe箔的WT图[77];(f) Na x LMNMT的Ni、Mn K边的原位XANES光谱[78];(g) 一系列Ti K边XANES,钛酸锂中Li嵌入过程中Li4/3Ti5/3O4 频谱加权函数随光谱指纹归一化幅度的演变以及锂化驱动的结构转变的演变[79](a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [71]; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle[73]; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM[75]; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration[76]; (e) WT plots of Fe- N3C2-C, Fe-N4-C, Fe2O3, and Fe foil[77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
... [73];(c) Air-NMM和Ar-NMM的Mn K边傅里叶变换EXAFS谱[75];(d) 由EXAFS分析得出的Ni-O和Mn-O配位数的变化和TM的迁移途径[76];(e) Fe-N3C2-C、Fe-N4-C、Fe2O3 和Fe箔的WT图[77];(f) Na x LMNMT的Ni、Mn K边的原位XANES光谱[78];(g) 一系列Ti K边XANES,钛酸锂中Li嵌入过程中Li4/3Ti5/3O4 频谱加权函数随光谱指纹归一化幅度的演变以及锂化驱动的结构转变的演变[79](a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [73]; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM[75]; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration[76]; (e) WT plots of Fe- N3C2-C, Fe-N4-C, Fe2O3, and Fe foil[77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [75]; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration[76]; (e) WT plots of Fe- N3C2-C, Fe-N4-C, Fe2O3, and Fe foil[77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [76]; (e) WT plots of Fe- N3C2-C, Fe-N4-C, Fe2O3, and Fe foil[77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [77]; (f) In situ XANES spectra at the Ni and Mn K-edge of Na x LMNMT[78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
... [78]; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li4/3Ti5/3O4 phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations[79]Fig. 4
(a) Normalized Mn K-edge XANES spectra of NMMO at different charged states<sup>[<xref ref-type="bibr" rid="R71">71</xref>]</sup>; (b) Mn L-edge XANES spectra and quantitative fitting results of NNMO and CTO@NNMO at the fully charged and discharged states of the 1st and 100th cycle<sup>[<xref ref-type="bibr" rid="R73">73</xref>]</sup>; (c) Mn K-edge Fourier-transformed EXAFS spectra of Air-NMM and Ar-NMM<sup>[<xref ref-type="bibr" rid="R75">75</xref>]</sup>; (d) Variation of Ni-O and Mn-O coordination numbers derived from EXAFS analysis and the pathway of TM migration<sup>[<xref ref-type="bibr" rid="R76">76</xref>]</sup>; (e) WT plots of Fe- N<sub>3</sub>C<sub>2</sub>-C, Fe-N<sub>4</sub>-C, Fe<sub>2</sub>O<sub>3</sub>, and Fe foil<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>; (f) <i>In situ</i> XANES spectra at the Ni and Mn K-edge of Na <i><sub>x</sub></i> LMNMT<sup>[<xref ref-type="bibr" rid="R78">78</xref>]</sup>; (g) Series of Ti K-edge XANES, Evolution of the spectral weight of Li<sub>4/3</sub>Ti<sub>5/3</sub>O<sub>4</sub> phase against normalized amplitude of spectral fingerprints during Li intercalation in lithium titanate and lithiation-driven structural transformations<sup>[<xref ref-type="bibr" rid="R79">79</xref>]</sup>Fig. 4
(a) Mn L<sub>3</sub>-edge RIXS maps and corresponding RIXS calculations on MnHCMn electrodes at the discharged and charged states<sup>[<xref ref-type="bibr" rid="R86">86</xref>]</sup>; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge<sup>[<xref ref-type="bibr" rid="R87">87</xref>]</sup>; (c) O K-edge RIXS maps of a Li<sub>1.2</sub>Ni<sub>0.2</sub>Mn<sub>0.6</sub>O<sub>2</sub> and Li<sub>1.2</sub>Ni<sub>0.2</sub>Ru<sub>0.6</sub>O<sub>2</sub> electrodes at different states of charge<sup>[<xref ref-type="bibr" rid="R88">88</xref>]</sup>; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge<sup>[<xref ref-type="bibr" rid="R84">84</xref>]</sup>Fig. 5
... 更重要的是,非原位状态表征难以清晰反映电池工况条件下的动态电子结构演变、离子及电子输运以及界面动态反应等关键信息,而原位XAS的成熟为探明电池充放电过程中真实的电荷补偿机制和复杂结构演变提供了可能.例如Guo等[78]利用Ni、Mn K边原位XANES以及EXAFS分析认为放电过程中P2-Na0.7Li0.03Mg0.03Ni0.27Mn0.6Ti0.07O2 (Na x LMNMT)中Mn未参与氧化还原反应,而Ni参与氧化还原反应并生成三价Ni进而导致J-T效应,使得晶体结构畸变[图4(f)].Sun等[85]通过对比第1圈与第451圈xLi2MnO3·(1-x)LiMeO2(Me=Mn,Ni,Co) (LMR-NMC)充放电过程中Mn K边的原位XANES谱图,发现450圈的充放电循环后Mn的氧化还原活性被极大地削弱了,并通过对Co、Ni K边原位XANES的分析,认为450圈后的充放电过程中Co失去大量活性,并且Ni已完全失活,因此将450圈后LMR-NMC的容量归因于阴离子氧化还原.另外,Wang等[79]通过对Ti K边原位XANES中边前峰积分强度、主峰位置等参数的分析,发现Li4/3Ti5/3O4在嵌锂过程中并非发生简单的两相相变,而是在亚晶胞和颗粒尺度上发生多阶段的结构转变[图4(g)].利用原位XAS分析能深入理解钠离子电池电极材料的电荷补偿机制与容量衰减机制,优化电池材料组成与结构,为进一步提升电池性能提供重要科学依据. ...
3
... RIXS技术能对不同激发能的发射光子进行分辨和统计,并进一步获得更高维度的信息,从而有效解决sXAS难以分辨的价态信息.例如,Wessells等[86]通过Mn L3边mRIXS谱图对Na x Mn[Mn(CN)6]0.81(MnHCMn)正极材料中Mn在充放电过程中的价态进行表征,发现放电过程中mRIXS谱图中出现多组平行于弹性峰的d-d激发特征区域,说明t2g和eg态被部分占据,证明了二价Mn的存在;充电过程中,mRIXS谱图在激发能为643.5 eV时具有较高的能量损失,这种特征对应于Mn1+低自旋3d6体系的d-d激发,反映该体系的t2g态被完全占据,从而证明了MnHCMn中一价Mn的存在[图5(a)].类似地,Yang等[49]通过对比发现,sXAS技术并不能区分一价Mn与二价Mn,这说明RIXS技术在价态分辨方面有不可替代的作用. ...
... [86];(b) 激发能在Mn L边、发射能在Mn L边和O K边初始态NNMO的mRIXS图像[87];(c) Li1.2Ni0.2Mn0.6O2 和Li1.2Ni0.2Ru0.6O2 电极在不同电荷状态下的O K边RIXS图[88];(d) NMMCO和NMMO在不同电荷状态下的O K边RIXS和sPFY光谱[84](a) Mn L<sub>3</sub>-edge RIXS maps and corresponding RIXS calculations on MnHCMn electrodes at the discharged and charged states<sup>[<xref ref-type="bibr" rid="R86">86</xref>]</sup>; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge<sup>[<xref ref-type="bibr" rid="R87">87</xref>]</sup>; (c) O K-edge RIXS maps of a Li<sub>1.2</sub>Ni<sub>0.2</sub>Mn<sub>0.6</sub>O<sub>2</sub> and Li<sub>1.2</sub>Ni<sub>0.2</sub>Ru<sub>0.6</sub>O<sub>2</sub> electrodes at different states of charge<sup>[<xref ref-type="bibr" rid="R88">88</xref>]</sup>; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge<sup>[<xref ref-type="bibr" rid="R84">84</xref>]</sup>Fig. 5
... [86]; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge[87]; (c) O K-edge RIXS maps of a Li1.2Ni0.2Mn0.6O2 and Li1.2Ni0.2Ru0.6O2 electrodes at different states of charge[88]; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge[84]Fig. 5
(a) Mn L<sub>3</sub>-edge RIXS maps and corresponding RIXS calculations on MnHCMn electrodes at the discharged and charged states<sup>[<xref ref-type="bibr" rid="R86">86</xref>]</sup>; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge<sup>[<xref ref-type="bibr" rid="R87">87</xref>]</sup>; (c) O K-edge RIXS maps of a Li<sub>1.2</sub>Ni<sub>0.2</sub>Mn<sub>0.6</sub>O<sub>2</sub> and Li<sub>1.2</sub>Ni<sub>0.2</sub>Ru<sub>0.6</sub>O<sub>2</sub> electrodes at different states of charge<sup>[<xref ref-type="bibr" rid="R88">88</xref>]</sup>; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge<sup>[<xref ref-type="bibr" rid="R84">84</xref>]</sup>Fig. 5
... [87]; (c) O K-edge RIXS maps of a Li1.2Ni0.2Mn0.6O2 and Li1.2Ni0.2Ru0.6O2 electrodes at different states of charge[88]; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge[84]Fig. 5
(a) Mn L<sub>3</sub>-edge RIXS maps and corresponding RIXS calculations on MnHCMn electrodes at the discharged and charged states<sup>[<xref ref-type="bibr" rid="R86">86</xref>]</sup>; (b) mRIXS images of pristine NNMO with excitation energy at Mn L-edge range, but with emission energy at Mn L-edge and O K-edge<sup>[<xref ref-type="bibr" rid="R87">87</xref>]</sup>; (c) O K-edge RIXS maps of a Li<sub>1.2</sub>Ni<sub>0.2</sub>Mn<sub>0.6</sub>O<sub>2</sub> and Li<sub>1.2</sub>Ni<sub>0.2</sub>Ru<sub>0.6</sub>O<sub>2</sub> electrodes at different states of charge<sup>[<xref ref-type="bibr" rid="R88">88</xref>]</sup>; (d) O K-edge RIXS and O-K sPFY spectra of NMMCO and NMMO at different states of charge<sup>[<xref ref-type="bibr" rid="R84">84</xref>]</sup>Fig. 5




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}