Energy and the environment are important pillars behind the sustainable development of human society. Therefore, the future society requires efficient, economical, green, and safe electrochemical energy storage field to deal with global climate change and energy crisis. Thus, it is crucial to understand the electrochemical interface reaction mechanism to further guide the design of energy storage devices. Time-of-flight secondary ion mass spectrometry (ToF-SIMS), including ex situ and in situ methods, has emerged in recent energy electrochemical fields due to its ultra-high sensitivity, as well as temporal and spatial resolution. This review summarizes the latest developments and applications of ToF-SIMS in recent technologies of energy electrochemistry (e.g., lithium-ion, -sulfur, and lithium-oxygen batteries), with critically considering the technology-assisted revelation of the electrochemical reaction process and further design of better electrochemical energy storage systems. Finally, we also discussed the current challenges and future opportunities of ToF-SIMS, and advocated the widespread use of the technology to guide the design and innovation of future energy storage technologies.
Keywords:electrochemical energy storage technology
;
time-of-flight secondary ion mass spectrometry
;
interface reaction mechanism
ZHAO Zhiwei. Application of time-of-flight secondary ion mass spectrometry in lithium-based rechargeable batteries[J]. Energy Storage Science and Technology, 2022, 11(3): 781-794
为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破。为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能。然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13]。这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变。在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14]。通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解。随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19]。其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术。传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]。
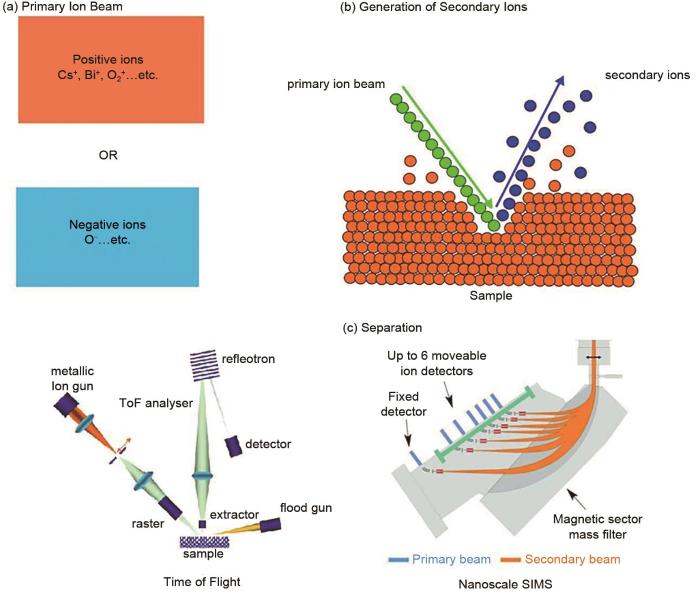
Fig. 1
Principles of secondary ion mass spectrometry (SIMS) analysis: (a) SIMS primary ion beam: positive ions or negative ions; (b) the primary ion beam strikes the sample surface during sputtering and then produces secondary ions; (c) secondary ions are accelerated towards the detector, either through a flight tube (ToF-SIMS) or through magnetic separation using a quadrupole (NanoSIMS)[23]
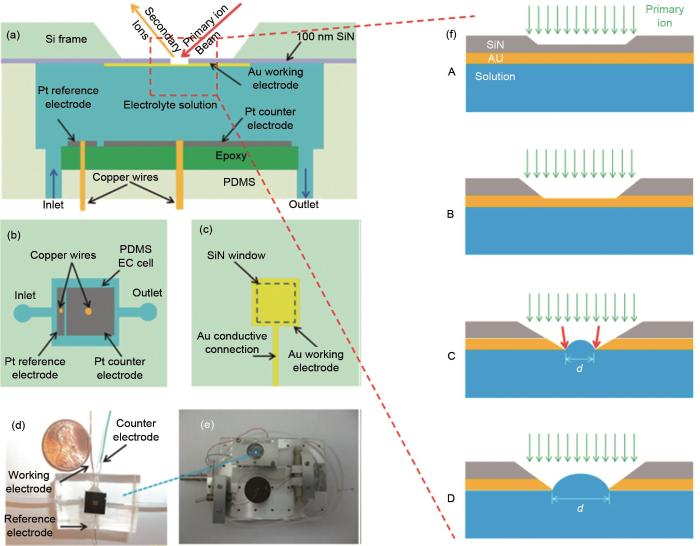
Fig. 2
The first in situ electrochemical ToF-SIMS analysis device, including (a) side view, (b) top view of the Pt counter electrode and reference electrode, (c) the top view of the Au working electrode, (d) the photograph of the device and (e) EC-cell assembly on the ToF-SIMS stage; (f) a schematic illustration of the aperture evolution during in situ measurements[26]
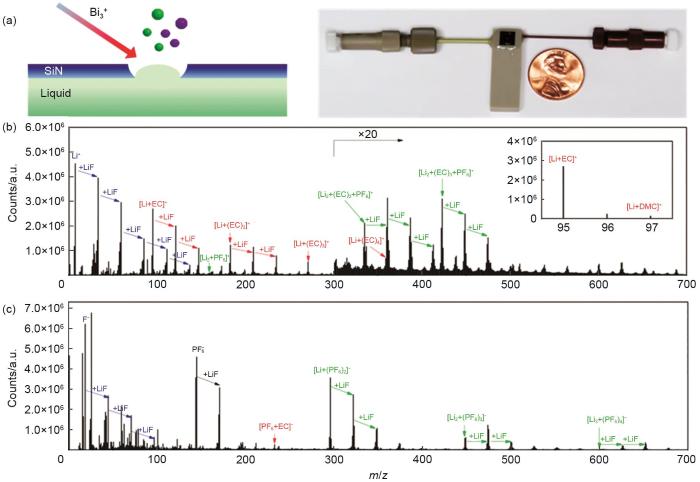
Fig. 3
(a) schematic illustration of SIMS measurement and the photograph of the liquid battery that is vacuum compatible; (b) positive ion spectra of 1.0 mol·L-1 LiPF6 in EC-DMC electrolyte; (c) negative ion spectra of 1.0 mol·L-1 LiPF6 in EC-DMC electrolyte[33]
Fig. 4
(a) schematic of the model cell; (b) a schematic illustration of in situ ToF-SIMS analysis of solid-liquid interface; (c) 3D reconstructions of the important SIMS signal on OCP state, charging to 2.0 V and discharged OCP state on Cu electrode surface[34]
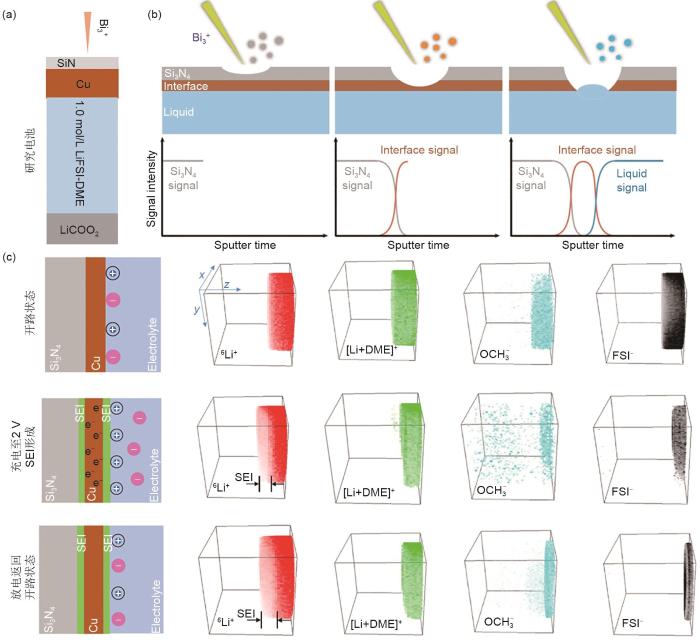
Fig. 5
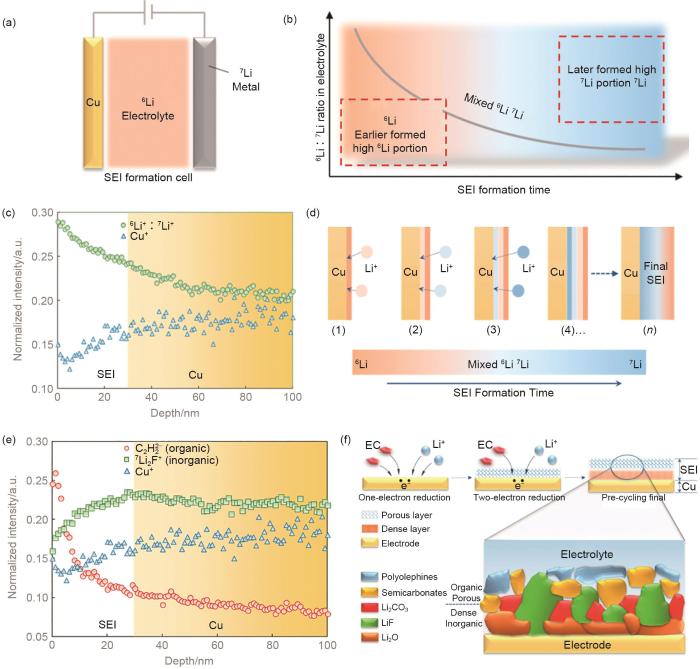
(a) schematic of a model cell; (b) Li isotope ratio variation trend over time in electrolyte during SEI formation process; (c) SIMS depth profiles of the 6Li∶ 7Li ratio and Cu+ of SEI on Cu electrode surface; (d) schematic of 6Li∶ 7Li isotope ratio variation over time on SEI of Cu electrode surface; (e) the depth profile of secondary ion of C2H2-(organic), Li2F-(inorganic) and Cu+ on Cu electrode; (f) schematic of SEI formation mechanism[37]
Fig. 6
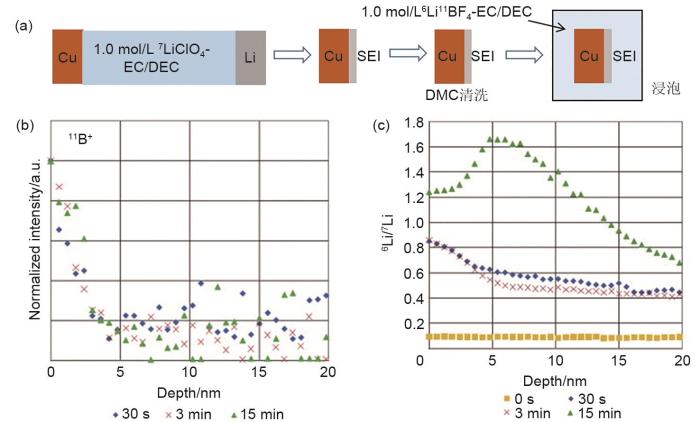
Schematic illustration of SEI treatment on Cu electrode surface; SEI formed in 7LiClO4-EC/DEC after immersion in 6LiBF4-EC/DEC electrolyte: (b) depth profile of secondary ion of 11B+ at various immersion time, and (c) 11B+ and 7Li2O+ at 3 min immersion time[38]
Fig. 7
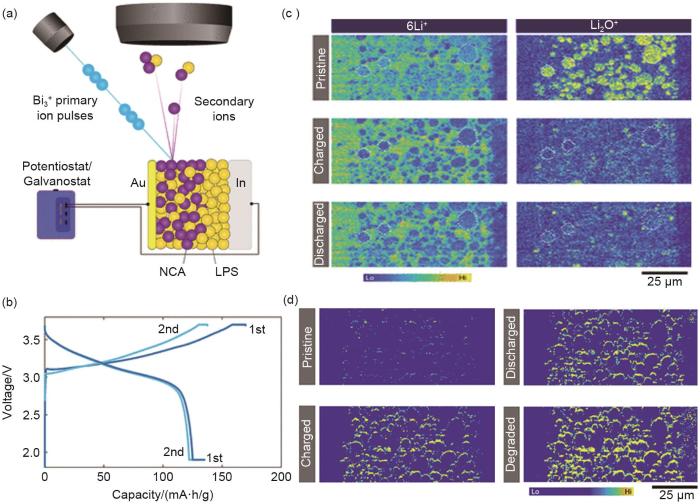
(a) schematic illustration of in situ ToF-SIMS measurement performed on an all-solid-state lithium-ion battery; (b) Charge and discharge profiles for the first and second cycle; (c) evolution of the distribution of 6Li+ and Li2O+ fragments during cycling; (d) evolution of the intensity of PO2- and PO3- secondary ion fragments during the cycling of the all-solid-state lithium-ion battery [42]
Fig. 8
(a) depth profiles of LiO-, LiS- and H- secondary ions that represents respectively Li2O, Li2S and various hydrogen-containing interphasial species, at 5, 40 and 300 cycles on lithium anode; (b) depth profiles of LiH-, LiOH- and C2H3- secondary ions that represents respectively LiH, LiOH and various organic interphasial species, at 5, 40, and 300 cycles on lithium anode; (c), (d) 3D reconstructions of the SIMS signal for LiH- and LiS- at 5, 40, and 300 cycles[45]
Fig. 9
3D distribution of secondary ion 18O- on the discharge electrode with ToF-SIMS depth scan: (a) 3D distribution of 18O- on the carbon (VC) electrode; (b) selected layers from the reconstructed 3D image of 18O- on the VC electrode at three depths; (c) 3D distribution of 18O- on the carbon load with Ru (Ru/VC) electrode; (d) selected layers from the reconstructed 3D image of 18O- on the Ru/VC electrode at three depths[48]
KWAK W J, ROSY, SHARON D, et al. Lithium-oxygen batteries and related systems: Potential, status, and future[J]. Chemical Reviews, 2020, 120(14): 6626-6683.
GAO H, GALLANT B M. Advances in the chemistry and applications of alkali-metal-gas batteries[J]. Nature Reviews Chemistry, 2020, 4(11): 566-583.
XU R, LU J, AMINE K. Progress in mechanistic understanding and characterization techniques of Li-S batteries[J]. Advanced Energy Materials, 2015, 5(16): doi: 10.1002/aenm.201500408.
LI X N, WANG H Y, YANG H B, et al. In situ/operando characterization techniques to probe the electrochemical reactions for energy conversion[J]. Small Methods, 2018, 2(6): doi: 10.1002/smtd.201700395.
LI H Y, GUO S H, ZHOU H S. In-situ/operando characterization techniques in lithium-ion batteries and beyond[J]. Journal of Energy Chemistry, 2021, 59: 191-211.
CHENG X B, ZHANG R, ZHAO C Z, et al. A review of solid electrolyte interphases on lithium metal anode[J]. Advanced Science, 2015, 3(3): doi: 10.1002/advs.201500213.
BHATTACHARYYA R, KEY B, CHEN H, et al. In situ NMR observation of the formation of metallic lithium microstructures in lithium batteries[J]. Nature Materials, 2010, 9(6): 504-510.
HUANG J Y, ZHONG L, WANG C M, et al. In situ observation of the electrochemical lithiation of a single SnO2 nanowire electrode[J]. Science, 2010, 330(6010): 1515-1520.
LIU X, WANG D, LIU G, et al. Distinct charge dynamics in battery electrodes revealed by in situ and operando soft X-ray spectroscopy[J]. Nature Communications, 2013, 4: doi: 10.1038/ncomms3568.
EBNER M, MARONE F, STAMPANONI M, et al. Visualization and quantification of electrochemical and mechanical degradation in Li ion batteries[J]. Science, 2013, 342(6159): 716-720.
LIU H, STROBRIDGE F C, BORKIEWICZ O J, et al. Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes[J]. Science, 2014, 344(6191): doi: 10.1126/science.1252817.
ZHU Z H, ZHOU Y F, YAN P F, et al. In situ mass spectrometric determination of molecular structural evolution at the solid electrolyte interphase in lithium-ion batteries[J]. Nano Letters, 2015, 15(9): 6170-6176.
LU J S, HUA X, LONG Y T. Recent advances in real-time and in situ analysis of an electrode-electrolyte interface by mass spectrometry[J]. The Analyst, 2017, 142(5): 691-699.
BONNIN E A, RIZZOLI S O. Novel secondary ion mass spectrometry methods for the examination of metabolic effects at the cellular and subcellular levels[J]. Frontiers in Behavioral Neuroscience, 2020, 14: doi: 10.3389/fnbeh.2020.00124.
NUÑEZ J, RENSLOW R, CLIFF J B 3rd, et al. NanoSIMS for biological applications: Current practices and analyses[J]. Biointerphases, 2017, 13(3): doi: 10.1116/1.4993628.
NIE M Y, ABRAHAM D P, SEO D M, et al. Role of solution structure in solid electrolyte interphase formation on graphite with LiPF6 in propylene carbonate[J]. The Journal of Physical Chemistry C, 2013, 117(48): 25381-25389.
HE Y T, ZHANG Y H, YU P, et al. Ion association tailoring SEI composition for Li metal anode protection[J]. Journal of Energy Chemistry, 2020, 45: 1-6.
ZHOU L, CAO Z, WAHYUDI W, et al. Electrolyte engineering enables high stability and capacity alloying anodes for sodium and potassium ion batteries[J]. ACS Energy Letters, 2020, 5(3): 766-776.
BORODIN O, REN X M, VATAMANU J, et al. Modeling insight into battery electrolyte electrochemical stability and interfacial structure[J]. Accounts of Chemical Research, 2017, 50(12): 2886-2894.
CRESCE A, BORODIN O, XU K. Correlating Li+ solvation sheath structure with interphasial chemistry on graphite[J]. The Journal of Physical Chemistry C, 2012, 116(50): 26111-26117.
ZHANG Y Y, SU M, YU X F, et al. Investigation of ion-solvent interactions in nonaqueous electrolytes using in situ liquid SIMS[J]. Analytical Chemistry, 2018, 90(5): 3341-3348.
ZHOU Y, SU M, YU X, et al. Real-time mass spectrometric characterization of the solid-electrolyte interphase of a lithium-ion battery[J]. Nature Nanotechnology, 2020, 15(3): 224-230.
TAKENAKA N, SUZUKI Y, SAKAI H, et al. On electrolyte-dependent formation of solid electrolyte interphase film in lithium-ion batteries: Strong sensitivity to small structural difference of electrolyte molecules[J]. The Journal of Physical Chemistry C, 2014, 118(20): 10874-10882.
LIU Z, LU P, ZHANG Q L, et al. A bottom-up formation mechanism of solid electrolyte interphase revealed by isotope-assisted time-of-flight secondary ion mass spectrometry[J]. The Journal of Physical Chemistry Letters, 2018, 9(18): 5508-5514.
BIELEFELD A, WEBER D A, JANEK J. Microstructural modeling of composite cathodes for all-solid-state batteries[J]. The Journal of Physical Chemistry C, 2019, 123(3): 1626-1634.
SAKUDA A, TAKEUCHI T, KOBAYASHI H. Electrode morphology in all-solid-state lithium secondary batteries consisting of LiNi1/3Co1/3Mn1/3O2 and Li2S-P2S5 solid electrolytes[J]. Solid State Ionics, 2016, 285: 112-117.
YAMAGISHI Y, MORITA H, NOMURA Y, et al. Visualizing lithium distribution and degradation of composite electrodes in sulfide-based all-solid-state batteries using operando time-of-flight secondary ion mass spectrometry[J]. ACS Applied Materials & Interfaces, 2021, 13(1): 580-586.
CHEN S R, NIU C J, LEE H, et al. Critical parameters for evaluating coin cells and pouch cells of rechargeable Li-metal batteries[J]. Joule, 2019, 3(4): 1094-1105.
NANDA S, MANTHIRAM A. Lithium degradation in lithium-sulfur batteries: Insights into inventory depletion and interphasial evolution with cycling[J]. Energy & Environmental Science, 2020, 13(8): 2501-2514.
WANG J W, ZHANG Y L, GUO L M, et al. Identifying reactive sites and transport limitations of oxygen reactions in aprotic lithium-O2 batteries at the stage of sudden death[J]. Angewandte Chemie, 2016, 55(17): 5201-5205.
WANG Y, LU Y C. Isotopic labeling reveals active reaction interfaces for electrochemical oxidation of lithium peroxide[J]. Angewandte Chemie, 2019, 131(21): 7036-7040.
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
0
1
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
1
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
1
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
0
0
0
1
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
1
... 为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破.为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能.然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13].这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变.在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14].通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解.随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19].其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术.传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1 ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]. ...
... [23]Principles of secondary ion mass spectrometry (SIMS) analysis: (a) SIMS primary ion beam: positive ions or negative ions; (b) the primary ion beam strikes the sample surface during sputtering and then produces secondary ions; (c) secondary ions are accelerated towards the detector, either through a flight tube (ToF-SIMS) or through magnetic separation using a quadrupole (NanoSIMS)<sup>[<xref ref-type="bibr" rid="R23">23</xref>]</sup>Fig. 1
... [26]The first in situ electrochemical ToF-SIMS analysis device, including (a) side view, (b) top view of the Pt counter electrode and reference electrode, (c) the top view of the Au working electrode, (d) the photograph of the device and (e) EC-cell assembly on the ToF-SIMS stage; (f) a schematic illustration of the aperture evolution during in situ measurements<sup>[<xref ref-type="bibr" rid="R26">26</xref>]</sup>Fig. 22 应用介绍<strong>2.1</strong> 锂离子电池
... [33](a) schematic illustration of SIMS measurement and the photograph of the liquid battery that is vacuum compatible; (b) positive ion spectra of 1.0 mol·L<sup>-1</sup> LiPF6 in EC-DMC electrolyte; (c) negative ion spectra of 1.0 mol·L<sup>-1</sup> LiPF6 in EC-DMC electrolyte<sup>[<xref ref-type="bibr" rid="R33">33</xref>]</sup>Fig. 32.1.2 SEI形成过程
... [34](a) schematic of the model cell; (b) a schematic illustration of in situ ToF-SIMS analysis of solid-liquid interface; (c) 3D reconstructions of the important SIMS signal on OCP state, charging to 2.0 V and discharged OCP state on Cu electrode surface<sup>[<xref ref-type="bibr" rid="R34">34</xref>]</sup>Fig. 42.1.3 SEI生长过程
... [37]<strong>(a) schematic of a model cell; (b) Li isotope ratio variation trend over time in electrolyte during SEI formation process; (c) SIMS depth profiles of the <sup>6</sup>Li</strong>∶ <strong><sup>7</sup>Li ratio and Cu<sup>+</sup> of SEI on Cu electrode surface; (d) schematic of <sup>6</sup>Li</strong>∶ <strong><sup>7</sup>Li isotope ratio variation over time on SEI of Cu electrode surface; (e) the depth profile of secondary ion of C<sub>2</sub>H<sub>2</sub></strong><sup>-</sup><strong>(organic), Li<sub>2</sub>F</strong><sup>-</sup><strong>(inorganic) and Cu<sup>+</sup> on Cu electrode; (f) schematic of SEI formation mechanism</strong><sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup>Fig. 52.1.4 离子传输
... [38]Schematic illustration of SEI treatment on Cu electrode surface; SEI formed in <sup>7</sup>LiClO<sub>4</sub>-EC/DEC after immersion in <sup>6</sup>LiBF<sub>4</sub>-EC/DEC electrolyte: (b) depth profile of secondary ion of <sup>11</sup>B<sup>+</sup> at various immersion time, and (c) <sup>11</sup>B<sup>+</sup> and <sup>7</sup>Li<sub>2</sub>O<sup>+</sup> at 3 min immersion time<sup>[<xref ref-type="bibr" rid="R38">38</xref>]</sup>Fig. 62.1.5 全固态电池
... [42](a) schematic illustration of in situ ToF-SIMS measurement performed on an all-solid-state lithium-ion battery; (b) Charge and discharge profiles for the first and second cycle; (c) evolution of the distribution of <sup>6</sup>Li<sup>+</sup> and Li<sub>2</sub>O<sup>+</sup> fragments during cycling; (d) evolution of the intensity of PO<sub>2</sub><sup>-</sup> and PO<sub>3</sub><sup>-</sup> secondary ion fragments during the cycling of the all-solid-state lithium-ion battery <sup>[<xref ref-type="bibr" rid="R42">42</xref>]</sup>Fig. 7<strong>2.2</strong> 锂硫电池
... [45](a) depth profiles of LiO<sup>-</sup>, LiS<sup>-</sup> and H<sup>-</sup> secondary ions that represents respectively Li<sub>2</sub>O, Li<sub>2</sub>S and various hydrogen-containing interphasial species, at 5, 40 and 300 cycles on lithium anode; (b) depth profiles of LiH<sup>-</sup>, LiOH<sup>-</sup> and C<sub>2</sub>H<sub>3</sub><sup>-</sup> secondary ions that represents respectively LiH, LiOH and various organic interphasial species, at 5, 40, and 300 cycles on lithium anode; (c), (d) 3D reconstructions of the SIMS signal for LiH<sup>-</sup> and LiS<sup>-</sup> at 5, 40, and 300 cycles<sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup>Fig. 8<strong>2.3</strong> 锂氧电池
... [48]3D distribution of secondary ion <sup>18</sup>O<sup>-</sup> on the discharge electrode with ToF-SIMS depth scan: (a) 3D distribution of <sup>18</sup>O<sup>-</sup> on the carbon (VC) electrode; (b) selected layers from the reconstructed 3D image of <sup>18</sup>O<sup>-</sup> on the VC electrode at three depths; (c) 3D distribution of <sup>18</sup>O<sup>-</sup> on the carbon load with Ru (Ru/VC) electrode; (d) selected layers from the reconstructed 3D image of <sup>18</sup>O<sup>-</sup> on the Ru/VC electrode at three depths<sup>[<xref ref-type="bibr" rid="R48">48</xref>]</sup>Fig. 93 总结和展望






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}