Monte Carlo (MC) simulation, on the basis of probability and statistics theory, was proposed by Von Neumann et al. in 1940s. As an important numerical method, MC simulation has been used to investigate thermodynamic and kinetic properties of ionic conductors. However, there exists a large improvable space for the MC simulation in the calculation accuracy, simulation efficiency and simulation process automation. In this work, through systematic analysis of the Hamiltonian model in MC simulation (For example, based on bond-valence theory or cluster expansion, the configuration energy is given by fitting the neighbors interaction parameters that can always be obtained by first principles calculations of representative small supercells) and the evolution model (For example, configuration evolution based on the assumption of single-ion jump mode) of material structure, a set of MC simulation paradigms for analyzing the ion transport and phase transition characteristics of ionic conductors are extracted, and the corresponding semi-automatic simulation codes are given which can be used for predicting the respective dependences of the ionic conductivity and the occupancy of the migrated ions in the garnet-structured ionic conductor with the lithium ion concentration. To expand the application of MC simulation in the research of ionic conductor, we further analyze its applications in the typical thermodynamic and kinetic properties calculations of electrochemical energy storage materials which includes anode and cathode materials, electrolytes and the related interfaces, including the ionic diffusion problems, the distribution characteristics of migrated ions and the evolution of related interface. Finally, the current challenges faced by MC methods are prospected and the possible solutions are presented, including: ① Accurately capturing all possible events (such as single-ion hop and two-ion cooperation hop) and their descriptions (such as the Hamiltonian calculations); ② Searching for efficient algorithm to accurately find the evolution trajectory of the system; ③ Accurately obtaining the corresponding actual time in the MC simulation.
Keywords:Monte Carlo simulation
;
ionic conductors
;
ion transport properties
;
phase transition properties
;
crystal growth
LIU Jinping. Investigating thermodynamic and kinetic properties of ionic conductors via Monte Carlo simulation[J]. Energy Storage Science and Technology, 2022, 11(3): 878-896
明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3]。离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5]。高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络。同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等)。借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化。分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用。然而其内禀缺陷限制了MD适用于更广泛的应用场景。例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数。因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性。表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度。此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势。
MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出。到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34]。20世纪60年代,KMC模拟开始出现。与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究。进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1)。根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟。这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态。从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然。多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟。而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC)。这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示。基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍。
Fig. 2
Several two-dimensional lattice-gas model which used in MC simulation commonly: (a) two-dimensional square lattice-gas model; (b) two-dimensional hexagonal lattice-gas model; (c) random lattice-gas model (The purple spheres represent the ions at the lattice sites)
Fig. 3
Approximation of the energy in MC simulation: (a) calculation of the interaction energy between lithium ions and other ions in graphite structure; (b) schematic diagram of the transition barrier during the jump of the migrated ions; (c) different jump modes of migrated ions; (d) different transition barrier of different jump modes; (e) relation diagram between some Monte Carlo simulations
式中,m为总的位点数目;为第j个位点被占据所引起的能量变化;为最近邻离子间的相互作用能;和为前置系数(其值取决于结构中离子的排布状态)。如果位点“j”被占据,则,反之。如果位点“j”和位点“i”同时被占据,并且它们互为最近邻位点,则,反之。另外,由Metropolis等[55]提出的系统演变概率计算公式具有精度较高、可有效避免结构在演变过程中进入局域能量极小值等优点,所以在MC模拟中得到广泛应用(利用此算法的MC模拟也可以叫做Metropolis Monte Carlo,MTMC)。依照此算法,组态a到组态b的演变概率(为
Fig. 4
General steps of MC simulation, in which listed the calculation process of MC sample data. Blue box is the module required by dynamic simulation, orange box is the module required by thermodynamic simulation, green box is the common module of the both, and gray box represent modules that exist in other simulation(Here, particles include charged ions and other particles, which will be described as ions in ionic conductors)
Fig. 5
KMC simulation process and calculation cases: (a) schematic diagram of modeling and simulation process of Li7La3Zr2O12(LLZO) by KMC (The model construction method or calculation content depends on specific research questions. Here, the garnet structure Li7La3Zr2O12 is taken as an example, and the model is constructed by BVSE and CAVD method); (b) schematic diagram of KMC simulation combined with percolation simulation (KMC simulation provides the structure model, migrated ion arrangement and ion hop mode for percolation simulation, and percolation simulation is used to calculate accessible ions and effective carriers concentration); (c) the comparison of occupancy calculated by this KMC program with the experimental results; (d) the comparison of ionic conductivity calculated by this KMC program with the experimental results of LLZO
需要补充的是,上述程序的开发过程依托于对固态电解质材料的计算,其中相关的输入参数使用到了本团队前期开发的BVSE[60]与CAVD的相关代码[6]。这些输入参数包括骨架结构的离子输运通道,离子在相邻位点之间迁移的平均势垒等。图5(c)~(d)给出了部分计算与实验的结果对比,由图可知,计算得到的Li x La3Zr2O12的离子占据情况与电导率变化均与实验结果相符。
图7
MC模拟对正极材料相变与开路电压的分析:(a)MC模拟模型,上图为Li x FePO4 中Li的分布;下图为对应的能量图。绿色的球代表了锂离子[59];(b) 在不考虑电荷间作用能的情况下,Li离子沿着 a 轴的排布。如图所示,LiFePO4(LFP)和FePO4(FP)相之间形成了清晰的相边界,Li0.5FePO4 相收缩到一个有限的区域[59];(c) Li x FePO4 橄榄石纳米晶体在室温下恒电流放电过程的MC模拟。灰点代表活性颗粒中的锂原子,表面电流密度为0.5 A/m2。四组图分别为0 s:初始固溶体,0.0001 s:形成两个富锂相并结合在一起,0.00056 s:富锂相生长,10.81 s:贫锂相几乎完全被消耗;(d) 电池电压与活性材料中Li浓度(mol)的关系[64];(e) MC模拟计算出的Li/LiNi x Mn2-x O4 电池电压分布,分别为 x = 0.1、 x = 0.2、 x = 0.3、 x = 0.5[65];(f) 全电池(LiMn2O4 正极和碳负极)开路电位与放电期间正极材料中Ni占据率的关系[19]
Fig. 7
MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li x FePO4, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis a without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO4 and FePO4 phases, and the Li0.5FePO4 phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li x FePO4 olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m-2. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi x Mn2-x O4 battery calculated by MC, x = 0.1, x = 0.2, x = 0.3, x = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn2O4 anode and carbon cathode) and the Ni occupation of the cathode during discharge
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟。Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征。DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制。晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异。在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)]。为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒。通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)]。这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程。此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]。
图8
Li离子排布对Li离子迁移势垒的影响[(a)~(c)][11];(d)a-Si与c-Si中Li离子的扩散系数[11];(e) 通过MC模拟获得的Li x C6 的第一性原理相图。图中符号为:G为石墨,Ⅱ为Ⅱ级相,ⅡD为Ⅱ级无序相,Ⅰ为Ⅰ级相,ⅠD为Ⅰ级无序相,Ⅱ'为Ⅱ级相且Li离子组合为2×2[66]
Fig. 8
Effect of Li ions arrangement on the transition barrier [(a)-(c)]; (d) diffusion coefficient of Li ion in a-Si and c-Si; (e) first-principles phase diagram obtained from Monte Carlo simulations. The phase regions are denoted in the following way: G (graphite), Ⅱ (stage Ⅱ), ⅡD (disordered stage Ⅱ), Ⅰ (stage Ⅰ), ⅠD (disordered stage Ⅰ), and Ⅱ′ (stage Ⅱ with 2×2 Li ordering)
Fig. 9
Distribution of Li ions by MC simulation in graphite anode: (a) lithium/graphite intercalation compound model; (b) ionic conduction model in graphite; (c) the relationship between Li ion content and time in the process of Li ion insertion and removal; (d) Rüdorff-Hoffmann model of lithium/graphite intercalation compound; (e) dumas-Herold lithium/graphite intercalation compound model; (f) changes in energy values corresponding to the two models
Fig. 10
(a) schematic diagram of transition barrier between Cr ion and Li ions; (b) schematic diagram of lithium ions separated by Cr ions in each one-dimensional channel; (c) variation relation of the simulated capacity with the dopant amount and super-cell size. The numbers 200, 300, 500, 1000 and 2000 refer to the size of the super-cell being used
对于固态电解质,其离子输运的动力学特性更加受到关注,KMC在其中的应用更为广泛。由于典型的固态电解质存在三大特点[68]:①相邻位点之间的离子迁移容易发生;②可供迁移离子占据的位点的数量大于迁移离子的数量;③这些可用的位点连接成一个连续的扩散通路。这些特点使得格子气KMC可以很好地适用于固态电解质材料的模拟。Morgan等[20]根据石榴石构型固态电解质的结构特征[图11(a)~(c)],采用晶格气体MC模拟预测了石榴石结构固态电解质Li x La3Zr2O12的相关系数变化趋势,发现当xLi = 3(来自位置能量)和xLi = 6(来自最近邻斥力)时,其存在特别强的相关效应[图11(d)~(e)]。
Fig. 11
Schematic diagram of ion diffusion network in garnet frame and the change of correlation coefficient with Li ion concentration: (a) three-dimensional drawings of ion transport channels connected by ZrO6 octahedron and LaO8 dodecahedron in garnet structures. Migrated ions are randomly distributed in tetrahedral and octahedral spaces. The interconnected tetrahedral gap and octahedral gap form a three-dimensional ion transport network; (b) schematic diagram of partial ring structure, in which tetrahedral and octahedral vacancies are partially occupied by lithium ions. It should be noted that not all vacancies need to be occupied by lithium ions; (c) two-dimensional geometric connections of garnet ion transport channels. One octahedral gap is connected to two tetrahedral gaps, and one tetrahedral gap is connected to four octahedral gaps (four octahedral gaps are connected to each face of the tetrahedron, two of which are not drawn in the two-dimensional topology). In (a), (b) and (c), octahedral vacancies are represented by blue and tetrahedral vacancies by yellow; (d) the change of collective correlation coefficient with Li ion concentration (xLi); (e) change of autocorrelation coefficient with Li ion concentration (xLi), xLi refers to the stoichiometric number of Li ions in Li x La3Zr2O12, ranging from 0 to 9
考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)]。由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确。为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算。他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒。这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量。作者进一步给出了这一研究范式在Li x CoO2中的应用。结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)]。通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制。此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级。综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的。
Fig. 12
Comparison of several KMC calculation processes: (a) schematic diagram of channel connection and ionic conductivity of LLZO only considering geometric effects; (b) ionic transport channel calculated by BVSE and the comparison of ion conductivity calculated by KMC with experimental observation. Solid blue curves and filled circles represent the ion conductivity simulated by KMC. The solid red line represents the ion conductivity effectively calculated (from the KMC simulation at high temperatures) and the dashed line represents the inferred ionic conductivity at lower temperatures. The triangular green curve represents the experimental observation conductivity; (c) several typical ion distribution states and the ion diffusion coefficient calculated by KMC based on cluster expansion[46]
图13
(a)~(g)为简单立方晶格表面不同配位下的晶体生长过程[70];(h) 晶体尺寸与溶液pH值与氨含量依赖关系的相图。图中显示了在粒径分布为高斯分布的情况下,溶液的平均粒径及其对应的一阶标准差。这里显示的所有数据都是从计算模型中获取的。绿色区域表示次级颗粒较小,黄色区域表示次级颗粒较大。浅蓝点的二次粒径分布标准差较小,紫色点的二次粒径分布标准差较大。在pH值和氨含量方面,沉积较大(Dpart约8 μm)和较小(Dpart约4 μm)的二次活性颗粒的最佳操作条件也在图中突出显示(用红色圆圈表示)[71];(i) T = 1100 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(j) T = 1400 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(k) SEI膜的生成过程,一般分为吸附、解吸、表面扩散和钝化等步骤;(l)SEI膜厚度、充电速度与温度的依赖关系[73]
Fig.13
(a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (Dpart ~8 μm) and smaller (Dpart ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate F =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate F =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperature
GROSJEAN C, MIRANDA P H, PERRIN M, et al. Assessment of world lithium resources and consequences of their geographic distribution on the expected development of the electric vehicle industry[J]. Renewable and Sustainable Energy Reviews, 2012, 16(3): 1735-1744.
LU X, LI H. Fundamental scientific aspects of lithium batteries (Ⅱ)—Defect chemistry in battery materials[J]. Energy Storage Science and Technology, 2013, 2(2): 157-164.
FLURI A, MARCOLONGO A, RODDATIS V, et al. Enhanced proton conductivity in Y-doped BaZrO3 via strain engineering[J]. Advanced Science, 2017, 4(12): doi: 10.1002/advs.201700467.
YAN J, WANG Q, WEI T, et al. Supercapacitors: Recent advances in design and fabrication of electrochemical supercapacitors with high energy densities[J]. Advanced Energy Materials, 2014, 4(4): doi: 10.1002/aenm.201300816.
MANTHIRAM A, YU X, WANG S. Lithium battery chemistries enabled by solid-state electrolytes[J]. Nature Reviews Materials, 2017, 2: doi: 10.1038/natrevmats.2016.103.
HE B, YE A, CHI S, et al. CAVD, towards better characterization of void space for ionic transport analysis[J]. Scientific Data, 2020, 7: doi: 10.1038/s41597-020-0491-x.
BLATOV V A, SHEVCHENKO A P, PROSERPIO D M. Applied topological analysis of crystal structures with the program package ToposPro[J]. Crystal Growth & Design, 2014, 14(7): 3576-3586.
CHEN H M, WONG L L, ADAMS S. SoftBV—A software tool for screening the materials genome of inorganic fast ion conductors[J]. Acta Crystallographica Section B, Structural Science, Crystal Engineering and Materials, 2019, 75(Pt 1): 18-33.
WONG L L, PHUAH K C, DAI R Y, et al. Bond valence pathway analyzer—An automatic rapid screening tool for fast ion conductors within softBV[J]. Chemistry of Materials, 2021, 33(2): 625-641.
ZOU Z Y, MA N, WANG A P, et al. Relationships between Na+ distribution, concerted migration, and diffusion properties in rhombohedral NASICON[J]. Advanced Energy Materials, 2020, 10(30): doi: 10.1002/aenm.202001486.
MOON J, LEE B, CHO M, et al. Ab initio and kinetic Monte Carlo simulation study of lithiation in crystalline and amorphous silicon[J]. Journal of Power Sources, 2014, 272: 1010-1017.
DE KLERK N J J, VAN DER MAAS E, WAGEMAKER M. Analysis of diffusion in solid-state electrolytes through MD simulations, improvement of the Li-ion conductivity in β-Li3PS4 as an example[J]. ACS Applied Energy Materials, 2018, 1(7): 3230-3242.
ZHU P P, SMITH R W. Dynamic simulation of crystal growth by Monte Carlo method—I. Model description and kinetics[J]. Acta Metallurgica et Materialia, 1992, 40(4): 683-692.
LYU D, WANG W, LIU J P, et al. Phase diagrams and magnetic properties of a ferrimagnetic Ising bilayer superlattice: A Monte Carlo study[J]. Journal of Magnetism and Magnetic Materials, 2018, 465: 348-359.
KAR P, HARINIPRIYA S. Modeling of lithium ion batteries employing grand canonical Monte Carlo and multiscale simulation[J]. Journal of the Electrochemical Society, 2014, 161(5): A726-A735.
MORGAN B J. Lattice-geometry effects in garnet solid electrolytes: A lattice-gas Monte Carlo simulation study[J]. Royal Society Open Science, 2017, 4(11): doi: 10.1098/rsos.170824.
WANG W, BI J L, LIU R J, et al. Effects of the single-ion anisotropy on magnetic and thermodynamic properties of a ferrimagnetic mixed-spin (1, 3/2) cylindrical Ising nanowire[J]. Superlattices and Microstructures, 2016, 98: 433-447.
MALIK R, ZHOU F, CEDER G. Phase diagram and electrochemical properties of mixed olivines from first-principles calculations[J]. Physical Review B, 2009, 79(21): doi: 10.1103/PhysRevB.79.214201.
RAN Y, ZOU Z, LIU B, et al. Towards prediction of ordered phases in rechargeable battery chemistry via group-subgroup transformation[J]. npj Computational Materials, 2021, 7: 1-11.
KIM J H, MYUNG S T, YOON C S, et al. Comparative study of LiNi0.5Mn1.5O4-δ and LiNi0.5Mn1.5O4 cathodes having two crystallographic structures: Fd3̄m and P4332[J]. Chemistry of Materials, 2004, 16(5): 906-914.
WU H, PI J C, LIU Q, et al. All-inorganic lead free double perovskite Li-battery anode material hosting high Li+ ion concentrations[J]. The Journal of Physical Chemistry Letters, 2021, 12(17): 4125-4129.
DARLING R, NEWMAN J. Dynamic Monte Carlo simulations of diffusion in LiyMn2O4[J]. Journal of the Electrochemical Society, 1999, 146(10): 3765-3772.
SHIMIZU A, TACHIKAWA H. Dynamics behavior of lithium in graphite lattice: MD calculation approach[J]. Journal of Physics and Chemistry of Solids, 2000, 61(12): 1895-1899.
DUANE S, KENNEDY A D, PENDLETON B J, et al. Hybrid Monte Carlo[J]. Physics Letters B, 1987, 195(2): 216-222.
HARDY J, DE PAZZIS O, POMEAU Y. Molecular dynamics of a classical lattice gas: Transport properties and time correlation functions[J]. Physical Review A, 1976, 13(5): 1949-1961.
BROWNE C B, POWLEY E, WHITEHOUSE D, et al. A survey of Monte Carlo tree search methods[J]. IEEE Transactions on Computational Intelligence and AI in Games, 2012, 4(1): 1-43.
ZSIGMOND G, LIEUTENANT K, MEZEI F. Monte Carlo simulations of neutron scattering instruments by VITESS: Virtual instrumentation tool for ESS[J]. Neutron News, 2002, 13(4): 11-14.
FRANCIS Z, INCERTI S, ZEIN S A, et al. Monte Carlo simulation of SARS-CoV-2 radiation-induced inactivation for vaccine development[J]. Radiation Research, 2021, 195(3): 221-229.
TURCHIN V F. On the computation of multidimensional integrals by the Monte-Carlo method[J]. Theory of Probability & Its Applications, 1971, 16(4): 720-724.
YANG Z P, MING J, QIU C X, et al. A multigrid multilevel Monte Carlo method for stokes-darcy model with random hydraulic conductivity and beavers-Joseph condition[J]. Journal of Scientific Computing, 2022, 90(2): 1-30.
CHENG J R, YUAN X H, ZHAO L, et al. GCMC simulation of hydrogen physisorption on carbon nanotubes and nanotube arrays[J]. Carbon, 2004, 42(10): 2019-2024.
COSOLI P, FERRONE M, PRICL S, et al. Hydrogen sulphide removal from biogas by zeolite adsorption: Part I. GCMC molecular simulations[J]. Chemical Engineering Journal, 2008, 145(1): 86-92.
DO D D, DO H D. Pore characterization of carbonaceous materials by DFT and GCMC simulations: A review[J]. Adsorption Science & Technology, 2003, 21(5): 389-423.
GAVILÁN-ARRIAZU E M, PINTO O A, LÓPEZ M B, et al. The kinetic origin of the Daumas-Hérold model for the Li-ion/graphite intercalation system[J]. Electrochemistry Communications, 2018, 93: 133-137.
DER V A, CEDER G, ASTA M, et al. First-principles theory of ionic diffusion with nondilute carriers[J]. Physical Review B, 2001, 64(18): doi: 10.1103/PhysRevB.64.184307.
METHEKAR R N, NORTHROP P W, CHEN K J, et al. Kinetic Monte Carlo simulation of surface heterogeneity in graphite anodes for lithium-ion batteries: Passive layer formation[C]//Proceedings of the 2011 American Control Conference, 2011: 1512-1517.
CHATTERJEE A, VLACHOS D G. An overview of spatial microscopic and accelerated kinetic Monte Carlo methods[J]. Journal of Computer-Aided Materials Design, 2007, 14(2): 253-308.
LEE J, URBAN A, LI X, et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries[J]. Science, 2014, 343(6170): 519-522.
WANG W, CHEN D D, LYU D, et al. Monte Carlo study of magnetic and thermodynamic properties of a ferrimagnetic Ising nanoparticle with hexagonal core-shell structure[J]. Journal of Physics and Chemistry of Solids, 2017, 108: 39-51.
MAAZI N, BOULECHFAR R. A modified grain growth Monte Carlo algorithm for increased calculation speed in the presence of Zener drag effect[J]. Materials Science and Engineering: B, 2019, 242: 52-62.
LONGONE P, MARTÍN Á, RAMIREZ-PASTOR A J. Lattice-gas Monte Carlo study of sI clathrate hydrates of ethylene: Stability analysis and cell distortion[J]. Fluid Phase Equilibria, 2020, 521: doi: 10.1016/j.fluid.2020.112739.
METROPOLIS N, ROSENBLUTH A W, ROSENBLUTH M N, et al. Equation of state calculations by fast computing machines[J]. The Journal of Chemical Physics, 1953, 21(6): 1087-1092.
YANG Y, WANG W, LYU D, et al. Monte Carlo study of magnetic behaviors in a quadrangle ferrimagnetic Ising nanoisland[J]. Journal of Physics and Chemistry of Solids, 2018, 120: 109-122.
GRIESHAMMER S, EISELE S. Kinetic Monte Carlo simulations for solid state ionics: Case studies with the MOCASSIN program[J]. Diffusion Foundations, 2021, 29: 117-142.
HE B, MI P H, YE A J, et al. A highly efficient and informative method to identify ion transport networks in fast ion conductors[J]. Acta Materialia, 2021, 203: doi: 10.1016/j.actamat.2020.116490.
THANGADURAI V, NARAYANAN S, PINZARU D. Garnet-type solid-state fast Li ion conductors for Li batteries: Critical review[J]. Chemical Society Reviews, 2014, 43(13): 4714-4727.
ETACHERI V, MAROM R, ELAZARI R, et al. Challenges in the development of advanced Li-ion batteries: A review[J]. Energy & Environmental Science, 2011, 4(9): doi: 10.1039/C1EE01598B.
YAMADA A, CHUNG S C, HINOKUMA K. Optimized LiFePO4 for lithium battery cathodes[J]. Journal of the Electrochemical Society, 2001, 148(3): doi: 10.1149/1.1348257.
HIN C. Kinetic Monte Carlo simulations of anisotropic lithium intercalation into LixFePO4 electrode nanocrystals[J]. Advanced Functional Materials, 2011, 21(13): 2477-2487.
ZHENG T, DAHN J R. Lattice-gas model to understand voltage profiles of LiNixMn2–xO4/Lielectrochemical cells[J]. Physical Review B, 1997, 56(7): 3800-3805.
PERSSON K, HINUMA Y, MENG Y S, et al. Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations[J]. Physical Review B, 2010, 82(12): doi: 10.1103/PhysRevB.82.125416.
OUYANG C Y, SHI S Q, WANG Z X, et al. The effect of Cr doping on Li ion diffusion in LiFePO4 from first principles investigations and Monte Carlo simulations[J]. Journal of Physics: Condensed Matterial, 2004, 16(13): 2265-2272.
HUANG C M, JOANNE C L, PATNAIK B S V, et al. Monte Carlo simulation of grain growth in polycrystalline materials[J]. Applied Surface Science, 2006, 252(11): 3997-4002.
BARAI P, FENG Z G, KONDO H, et al. Multiscale computational model for particle size evolution during coprecipitation of Li-ion battery cathode precursors[J]. The Journal of Physical Chemistry B, 2019, 123(15): 3291-3303.
CHEN X J, ZHAO H, AI W S. Study on the competitive growth mechanism of SiC polytypes using Kinetic Monte Carlo method[J]. Journal of Crystal Growth, 2021, 559: doi: 10.1016/j.jcrysgro. 2021.126042.
HAO F, LIU Z X, BALBUENA P B, et al. Mesoscale elucidation of solid electrolyte interphase layer formation in Li-ion battery anode[J]. The Journal of Physical Chemistry C, 2017, 121(47): 26233-26240.
HUANG J, ZHONG P, HA Y, et al. Non-topotactic reactions enable high rate capability in Li-rich cathode materials[J]. Nature Energy, 2021, 6(7): 706-714.
SHI S Q, GAO J, LIU Y, et al. Multi-scale computation methods: Their applications in lithium-ion battery research and development[J]. Chinese Physics B, 2016, 25(1): doi: 10.1088/1674-1056/25/1/018212.
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
0
0
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
2
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... 需要补充的是,上述程序的开发过程依托于对固态电解质材料的计算,其中相关的输入参数使用到了本团队前期开发的BVSE[60]与CAVD的相关代码[6].这些输入参数包括骨架结构的离子输运通道,离子在相邻位点之间迁移的平均势垒等.图5(c)~(d)给出了部分计算与实验的结果对比,由图可知,计算得到的Li x La3Zr2O12的离子占据情况与电导率变化均与实验结果相符. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
4
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... [8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... 考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)].由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确.为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算.他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒.这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量.作者进一步给出了这一研究范式在Li x CoO2中的应用.结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)].通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制.此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级.综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的. ...
... [8];(c) 几种典型的离子排布状态与基于局域结构团簇展开的KMC计算得到的离子扩散系数[46]Comparison of several KMC calculation processes: (a) schematic diagram of channel connection and ionic conductivity of LLZO only considering geometric effects; (b) ionic transport channel calculated by BVSE and the comparison of ion conductivity calculated by KMC with experimental observation. Solid blue curves and filled circles represent the ion conductivity simulated by KMC. The solid red line represents the ion conductivity effectively calculated (from the KMC simulation at high temperatures) and the dashed line represents the inferred ionic conductivity at lower temperatures. The triangular green curve represents the experimental observation conductivity; (c) several typical ion distribution states and the ion diffusion coefficient calculated by KMC based on cluster expansion<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup>Fig. 12
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
2
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... , 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
4
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... 除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
... [11];(d)a-Si与c-Si中Li离子的扩散系数[11];(e) 通过MC模拟获得的Li x C6 的第一性原理相图.图中符号为:G为石墨,Ⅱ为Ⅱ级相,ⅡD为Ⅱ级无序相,Ⅰ为Ⅰ级相,ⅠD为Ⅰ级无序相,Ⅱ'为Ⅱ级相且Li离子组合为2×2[66]Effect of Li ions arrangement on the transition barrier [(a)-(c)]; (d) diffusion coefficient of Li ion in a-Si and c-Si; (e) first-principles phase diagram obtained from Monte Carlo simulations. The phase regions are denoted in the following way: G (graphite), Ⅱ (stage Ⅱ), ⅡD (disordered stage Ⅱ), Ⅰ (stage Ⅰ), ⅠD (disordered stage Ⅰ), and Ⅱ′ (stage Ⅱ with 2×2 Li ordering)Fig. 8
... [11];(e) 通过MC模拟获得的Li x C6 的第一性原理相图.图中符号为:G为石墨,Ⅱ为Ⅱ级相,ⅡD为Ⅱ级无序相,Ⅰ为Ⅰ级相,ⅠD为Ⅰ级无序相,Ⅱ'为Ⅱ级相且Li离子组合为2×2[66]Effect of Li ions arrangement on the transition barrier [(a)-(c)]; (d) diffusion coefficient of Li ion in a-Si and c-Si; (e) first-principles phase diagram obtained from Monte Carlo simulations. The phase regions are denoted in the following way: G (graphite), Ⅱ (stage Ⅱ), ⅡD (disordered stage Ⅱ), Ⅰ (stage Ⅰ), ⅠD (disordered stage Ⅰ), and Ⅱ′ (stage Ⅱ with 2×2 Li ordering)Fig. 8
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
3
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍.
Several two-dimensional lattice-gas model which used in MC simulation commonly: (a) two-dimensional square lattice-gas model; (b) two-dimensional hexagonal lattice-gas model; (c) random lattice-gas model (The purple spheres represent the ions at the lattice sites)Fig. 2
Approximation of the energy in MC simulation: (a) calculation of the interaction energy between lithium ions and other ions in graphite structure; (b) schematic diagram of the transition barrier during the jump of the migrated ions; (c) different jump modes of migrated ions; (d) different transition barrier of different jump modes; (e) relation diagram between some Monte Carlo simulationsFig. 3<strong>1.2</strong> 蒙特卡罗模拟过程
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
4
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... MC模拟与MD模拟的对比[18-20, 28-32] ...
... Comparison between MC simulation and MD simulation[18-20, 28-32] ...
<strong>MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li <i><sub>x</sub></i> FePO<sub>4</sub>, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis <i>a</i> without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO<sub>4</sub> and FePO<sub>4</sub> phases, and the Li<sub>0.5</sub>FePO<sub>4</sub> phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li <i><sub>x</sub></i> FePO<sub>4</sub> olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m<sup>-2</sup>. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi <i><sub>x</sub></i> Mn<sub>2</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> O<sub>4</sub> battery calculated by MC, <i>x</i> = 0.1, <i>x</i> = 0.2, <i>x</i> = 0.3, <i>x</i> = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn<sub>2</sub>O<sub>4</sub> anode and carbon cathode) and the Ni occupation of the cathode during discharge</strong>Fig. 7
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
9
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... MC模拟与MD模拟的对比[18-20, 28-32] ...
... Comparison between MC simulation and MD simulation[18-20, 28-32] ...
... 对于固态电解质,其离子输运的动力学特性更加受到关注,KMC在其中的应用更为广泛.由于典型的固态电解质存在三大特点[68]:①相邻位点之间的离子迁移容易发生;②可供迁移离子占据的位点的数量大于迁移离子的数量;③这些可用的位点连接成一个连续的扩散通路.这些特点使得格子气KMC可以很好地适用于固态电解质材料的模拟.Morgan等[20]根据石榴石构型固态电解质的结构特征[图11(a)~(c)],采用晶格气体MC模拟预测了石榴石结构固态电解质Li x La3Zr2O12的相关系数变化趋势,发现当xLi = 3(来自位置能量)和xLi = 6(来自最近邻斥力)时,其存在特别强的相关效应[图11(d)~(e)]. ...
... [20];(e) 自相关系数随着Li离子浓度(xLi)的变化[20], xLi 指的是Li x La3Zr2O12 中Li离子的化学计量数,取值0~9Schematic diagram of ion diffusion network in garnet frame and the change of correlation coefficient with Li ion concentration: (a) three-dimensional drawings of ion transport channels connected by ZrO<sub>6</sub> octahedron and LaO<sub>8</sub> dodecahedron in garnet structures. Migrated ions are randomly distributed in tetrahedral and octahedral spaces. The interconnected tetrahedral gap and octahedral gap form a three-dimensional ion transport network; (b) schematic diagram of partial ring structure, in which tetrahedral and octahedral vacancies are partially occupied by lithium ions. It should be noted that not all vacancies need to be occupied by lithium ions; (c) two-dimensional geometric connections of garnet ion transport channels. One octahedral gap is connected to two tetrahedral gaps, and one tetrahedral gap is connected to four octahedral gaps (four octahedral gaps are connected to each face of the tetrahedron, two of which are not drawn in the two-dimensional topology). In (a), (b) and (c), octahedral vacancies are represented by blue and tetrahedral vacancies by yellow; (d) the change of collective correlation coefficient with Li ion concentration (<i>x</i><sub>Li</sub>); (e) change of autocorrelation coefficient with Li ion concentration (<i>x</i><sub>Li</sub>), <i>x</i><sub>Li</sub> refers to the stoichiometric number of Li ions in Li <i><sub>x</sub></i> La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub>, ranging from 0 to 9Fig. 11
考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)].由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确.为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算.他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒.这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量.作者进一步给出了这一研究范式在Li x CoO2中的应用.结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)].通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制.此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级.综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的. ...
... [20], xLi 指的是Li x La3Zr2O12 中Li离子的化学计量数,取值0~9Schematic diagram of ion diffusion network in garnet frame and the change of correlation coefficient with Li ion concentration: (a) three-dimensional drawings of ion transport channels connected by ZrO<sub>6</sub> octahedron and LaO<sub>8</sub> dodecahedron in garnet structures. Migrated ions are randomly distributed in tetrahedral and octahedral spaces. The interconnected tetrahedral gap and octahedral gap form a three-dimensional ion transport network; (b) schematic diagram of partial ring structure, in which tetrahedral and octahedral vacancies are partially occupied by lithium ions. It should be noted that not all vacancies need to be occupied by lithium ions; (c) two-dimensional geometric connections of garnet ion transport channels. One octahedral gap is connected to two tetrahedral gaps, and one tetrahedral gap is connected to four octahedral gaps (four octahedral gaps are connected to each face of the tetrahedron, two of which are not drawn in the two-dimensional topology). In (a), (b) and (c), octahedral vacancies are represented by blue and tetrahedral vacancies by yellow; (d) the change of collective correlation coefficient with Li ion concentration (<i>x</i><sub>Li</sub>); (e) change of autocorrelation coefficient with Li ion concentration (<i>x</i><sub>Li</sub>), <i>x</i><sub>Li</sub> refers to the stoichiometric number of Li ions in Li <i><sub>x</sub></i> La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub>, ranging from 0 to 9Fig. 11
考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)].由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确.为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算.他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒.这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量.作者进一步给出了这一研究范式在Li x CoO2中的应用.结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)].通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制.此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级.综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的. ...
... 考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)].由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确.为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算.他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒.这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量.作者进一步给出了这一研究范式在Li x CoO2中的应用.结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)].通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制.此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级.综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的.
Comparison of several KMC calculation processes: (a) schematic diagram of channel connection and ionic conductivity of LLZO only considering geometric effects; (b) ionic transport channel calculated by BVSE and the comparison of ion conductivity calculated by KMC with experimental observation. Solid blue curves and filled circles represent the ion conductivity simulated by KMC. The solid red line represents the ion conductivity effectively calculated (from the KMC simulation at high temperatures) and the dashed line represents the inferred ionic conductivity at lower temperatures. The triangular green curve represents the experimental observation conductivity; (c) several typical ion distribution states and the ion diffusion coefficient calculated by KMC based on cluster expansion<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup>Fig. 12
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
1
... 明确离子导体的离子输运与相变特性是制备新型全固态电池、电化学转换器件等离子型器件的关键[1-3].离子导体的典型结构包括在输运网络中嵌入或迁移实现电荷储存与传输的迁移离子以及构建离子输运网络的仅原点附近振动的骨架离子[4-5].高离子电导率的超离子导体的筛选过程通常采用高通量计算方法[如几何分析方法(crystal structure analysis by voronoi decomposition,CAVD)[6-7]、配位成键理论(bond valence site energy,BVSE)[8-9]等]分析骨架离子中的离子输运网络.同时,借助基于牛顿力学的分子动力学(molecular dynamics,MD)模拟[10]或基于离子迁移实验的动力学蒙特卡罗(kinetic Monte Carlo,KMC)模拟[11]分析迁移离子的输运特性(例如离子电导率[12-13]、扩散系数[8, 10, 14]等).借助群论、巨正则蒙特卡罗(grand canonical Monte Carlo,GCMC)模拟等热力学模拟手段研究离子导体的结构演化.分子动力学(MD)模拟作为典型的动力学模拟手段因其计算精度高、模拟分子运动过程清晰等优势,在离子扩散模式分析[15]及晶体生长模拟[16-17]等方面具有广泛应用.然而其内禀缺陷限制了MD适用于更广泛的应用场景.例如,第一性原理分子动力学模拟计算速度较慢、难以应用到较大体系的快速模拟;而平均场分子动力学模拟不易获取其所需的高精度势函数.因此,具有类似计算功能的KMC模拟因计算速度快、扩展性强而被广泛应用于评估离子导体的离子输运特性.表1对几种典型的MD模拟与MC模拟[18-20]进行了对比,尽管两者的应用范围存在众多交叠,但基于计算原理的差异性,MC模拟具有更广泛的时空尺度.此外,在热力学模拟领域(如相图计算[21]、电压平台计算等),相较于第一性原理相图计算[22]以及空间群分解[23]等模拟手段,MC模拟同样在计算速度与适用性等方面存在巨大优势. ...
... Comparison between MC simulation and MD simulation[18-20, 28-32] ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... [28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
4
... MC模拟与MD模拟的对比[18-20, 28-32] ...
... Comparison between MC simulation and MD simulation[18-20, 28-32] ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... , 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
2
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
0
0
0
1
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
1
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
1
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍. ...
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍.
Several two-dimensional lattice-gas model which used in MC simulation commonly: (a) two-dimensional square lattice-gas model; (b) two-dimensional hexagonal lattice-gas model; (c) random lattice-gas model (The purple spheres represent the ions at the lattice sites)Fig. 2
Approximation of the energy in MC simulation: (a) calculation of the interaction energy between lithium ions and other ions in graphite structure; (b) schematic diagram of the transition barrier during the jump of the migrated ions; (c) different jump modes of migrated ions; (d) different transition barrier of different jump modes; (e) relation diagram between some Monte Carlo simulationsFig. 3<strong>1.2</strong> 蒙特卡罗模拟过程
... [45]Distribution of Li ions by MC simulation in graphite anode: (a) lithium/graphite intercalation compound model; (b) ionic conduction model in graphite; (c) the relationship between Li ion content and time in the process of Li ion insertion and removal; (d) Rüdorff-Hoffmann model of lithium/graphite intercalation compound; (e) dumas-Herold lithium/graphite intercalation compound model; (f) changes in energy values corresponding to the two modelsFig. 9<strong>3.2</strong> 离子输运特性的蒙特卡罗模拟
... MC模拟作为一种基于随机抽样统计的模拟手段[33],由美国数学家冯·诺伊曼等[28]于20世纪40年代在美国的“曼哈顿计划”中提出.到20世纪50年代,进一步提出了MC模拟中可适用于大多数物理平衡系统的著名算法:Metropolis算法[34].20世纪60年代,KMC模拟开始出现.与此同时,混合蒙特卡罗(hybrid Monte Carlo,HMC)采样算法[31]等也得到了广泛地研究.进入21世纪以来,随着人工智能的快速崛起,蒙特卡罗树搜索算法等更为先进实用的计算方案也得到了快速发展[35-39](图1).根据研究对象的不同,MC模拟可分为两类:一类是针对热力学平衡过程中非时间关联性问题的模拟,如常见的GCMC模拟;另一类是对时间关联的动力学问题的研究,如KMC模拟.这两种MC模拟的最终目标都是尽可能地寻找徘徊在某一低势能面附近的结构组态.从原理上讲,GCMC与KMC之间存在应用场景的重合,即巨正则体系依旧可以进行KMC模拟,反之亦然.多数KMC模拟与热力学MC模拟均可以利用格子气[32, 40]模型建模,故而上述多数模拟均属于格子气蒙特卡罗(lattice-gas Monte Carlo,LGMC)模拟.而格子气模型的MC模拟多属于典型的马尔科夫链(Markov chain)[28, 33, 41]过程,所以这种MC模拟也可以叫作马尔科夫链蒙特卡罗(Markov chain Monte Carlo,MCMC).这几种模拟手段之间的关系可以通过图3(e)所示的维恩图表示.基于上述几种MC模拟的计算过程大多相似,本文将在格子气MC模拟的介绍中对体系描述、体系演变及新体系接受概率等细节问题进行重点介绍.
Several two-dimensional lattice-gas model which used in MC simulation commonly: (a) two-dimensional square lattice-gas model; (b) two-dimensional hexagonal lattice-gas model; (c) random lattice-gas model (The purple spheres represent the ions at the lattice sites)Fig. 2
Approximation of the energy in MC simulation: (a) calculation of the interaction energy between lithium ions and other ions in graphite structure; (b) schematic diagram of the transition barrier during the jump of the migrated ions; (c) different jump modes of migrated ions; (d) different transition barrier of different jump modes; (e) relation diagram between some Monte Carlo simulationsFig. 3<strong>1.2</strong> 蒙特卡罗模拟过程
... 考虑到键价和理论等静态计算方法的局限性,Chen等[8]提出将键价和理论计算的迁移能垒数据与KMC模拟相结合的方法,得到了指定温度下的绝对电导率[图12(b)],这显然优于仅考虑几何效应计算得到的电导率变化趋势[图12(a)].由于迁移离子的具体排布对离子迁移能垒具有极大的影响,所以上述的近似也不够精确.为此,Ven等[46]基于第一性原理团簇展开的方式,对KMC模拟的势垒参数进行了计算.他们发现在无序材料中,应用团簇展开可以近似获得任何构型下的迁移势垒.这意味着在KMC模拟中,任何时刻都能近似地保持细致平衡,并能在热力学平衡状态下计算出所需的动力学量.作者进一步给出了这一研究范式在Li x CoO2中的应用.结果表明,迁移机制和激活能垒在很大程度上取决于迁移锂离子周围近邻锂离子与锂空位的排布[图12(c)].通过模拟,作者发现在所有锂浓度下,锂在层状Li x CoO2中的扩散依赖于双空位机制.此外,由于激活能垒受离子浓度影响巨大,扩散系数随锂离子浓度x的变化可以达到几个数量级.综上所述,KMC的模拟一般依赖于其他计算方法的输入(包括模型结构,迁移能垒等),这对计算结果精度的影响是巨大的. ...
... [46]Comparison of several KMC calculation processes: (a) schematic diagram of channel connection and ionic conductivity of LLZO only considering geometric effects; (b) ionic transport channel calculated by BVSE and the comparison of ion conductivity calculated by KMC with experimental observation. Solid blue curves and filled circles represent the ion conductivity simulated by KMC. The solid red line represents the ion conductivity effectively calculated (from the KMC simulation at high temperatures) and the dashed line represents the inferred ionic conductivity at lower temperatures. The triangular green curve represents the experimental observation conductivity; (c) several typical ion distribution states and the ion diffusion coefficient calculated by KMC based on cluster expansion<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup>Fig. 12
... 式中,m为总的位点数目;为第j个位点被占据所引起的能量变化;为最近邻离子间的相互作用能;和为前置系数(其值取决于结构中离子的排布状态).如果位点“j”被占据,则,反之.如果位点“j”和位点“i”同时被占据,并且它们互为最近邻位点,则,反之.另外,由Metropolis等[55]提出的系统演变概率计算公式具有精度较高、可有效避免结构在演变过程中进入局域能量极小值等优点,所以在MC模拟中得到广泛应用(利用此算法的MC模拟也可以叫做Metropolis Monte Carlo,MTMC).依照此算法,组态a到组态b的演变概率(为 ...
<strong>MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li <i><sub>x</sub></i> FePO<sub>4</sub>, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis <i>a</i> without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO<sub>4</sub> and FePO<sub>4</sub> phases, and the Li<sub>0.5</sub>FePO<sub>4</sub> phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li <i><sub>x</sub></i> FePO<sub>4</sub> olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m<sup>-2</sup>. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi <i><sub>x</sub></i> Mn<sub>2</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> O<sub>4</sub> battery calculated by MC, <i>x</i> = 0.1, <i>x</i> = 0.2, <i>x</i> = 0.3, <i>x</i> = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn<sub>2</sub>O<sub>4</sub> anode and carbon cathode) and the Ni occupation of the cathode during discharge</strong>Fig. 7
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
... [59];(c) Li x FePO4 橄榄石纳米晶体在室温下恒电流放电过程的MC模拟.灰点代表活性颗粒中的锂原子,表面电流密度为0.5 A/m2.四组图分别为0 s:初始固溶体,0.0001 s:形成两个富锂相并结合在一起,0.00056 s:富锂相生长,10.81 s:贫锂相几乎完全被消耗;(d) 电池电压与活性材料中Li浓度(mol)的关系[64];(e) MC模拟计算出的Li/LiNi x Mn2-x O4 电池电压分布,分别为 x = 0.1、 x = 0.2、 x = 0.3、 x = 0.5[65];(f) 全电池(LiMn2O4 正极和碳负极)开路电位与放电期间正极材料中Ni占据率的关系[19]<strong>MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li <i><sub>x</sub></i> FePO<sub>4</sub>, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis <i>a</i> without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO<sub>4</sub> and FePO<sub>4</sub> phases, and the Li<sub>0.5</sub>FePO<sub>4</sub> phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li <i><sub>x</sub></i> FePO<sub>4</sub> olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m<sup>-2</sup>. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi <i><sub>x</sub></i> Mn<sub>2</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> O<sub>4</sub> battery calculated by MC, <i>x</i> = 0.1, <i>x</i> = 0.2, <i>x</i> = 0.3, <i>x</i> = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn<sub>2</sub>O<sub>4</sub> anode and carbon cathode) and the Ni occupation of the cathode during discharge</strong>Fig. 7
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
1
... 需要补充的是,上述程序的开发过程依托于对固态电解质材料的计算,其中相关的输入参数使用到了本团队前期开发的BVSE[60]与CAVD的相关代码[6].这些输入参数包括骨架结构的离子输运通道,离子在相邻位点之间迁移的平均势垒等.图5(c)~(d)给出了部分计算与实验的结果对比,由图可知,计算得到的Li x La3Zr2O12的离子占据情况与电导率变化均与实验结果相符. ...
General steps of MC simulation, in which listed the calculation process of MC sample data. Blue box is the module required by dynamic simulation, orange box is the module required by thermodynamic simulation, green box is the common module of the both, and gray box represent modules that exist in other simulation(Here, particles include charged ions and other particles, which will be described as ions in ionic conductors)Fig. 4
KMC simulation process and calculation cases: (a) schematic diagram of modeling and simulation process of Li<sub>7</sub>La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub>(LLZO) by KMC (The model construction method or calculation content depends on specific research questions. Here, the garnet structure Li<sub>7</sub>La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub> is taken as an example, and the model is constructed by BVSE and CAVD method); (b) schematic diagram of KMC simulation combined with percolation simulation (KMC simulation provides the structure model, migrated ion arrangement and ion hop mode for percolation simulation, and percolation simulation is used to calculate accessible ions and effective carriers concentration); (c) the comparison of occupancy calculated by this KMC program with the experimental results; (d) the comparison of ionic conductivity calculated by this KMC program with the experimental results of LLZOFig. 5
MC模拟常用的输入与输出 ...
... [61]的比较KMC simulation process and calculation cases: (a) schematic diagram of modeling and simulation process of Li<sub>7</sub>La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub>(LLZO) by KMC (The model construction method or calculation content depends on specific research questions. Here, the garnet structure Li<sub>7</sub>La<sub>3</sub>Zr<sub>2</sub>O<sub>12</sub> is taken as an example, and the model is constructed by BVSE and CAVD method); (b) schematic diagram of KMC simulation combined with percolation simulation (KMC simulation provides the structure model, migrated ion arrangement and ion hop mode for percolation simulation, and percolation simulation is used to calculate accessible ions and effective carriers concentration); (c) the comparison of occupancy calculated by this KMC program with the experimental results; (d) the comparison of ionic conductivity calculated by this KMC program with the experimental results of LLZOFig. 5
MC模拟常用的输入与输出 ...
1
... 在锂离子电池中,正极材料的离子输运特性直接决定了电池的容量与循环性能[62].LiFePO4[63]作为一种被广泛商用的正极材料,在平衡状态下,会自发地分裂出贫锂和富锂两相.如何快速获取相的分离过程十分重要.Xiao等[59]通过DFT计算得到KMC模拟的能垒输入,搭建了如图7(a)所示的模型.模拟结果显示,LiFePO4和FePO4之间形成有序的Li0.5FePO4相[图7(b)].Hin等[64]通过KMC模拟了Li x FePO4橄榄石纳米晶体中锂离子脱出与嵌入动力学特性.结果显示,锂离子嵌入过程中电池电压的变化包含以下几个阶段:①锂离子插入贫锂相导致电池电势下降;②富锂相Li1-β FePO4成核后,电势几乎恒定;③贫锂相完全演变为富锂相以后,电池的电势继续下降[图7(c)~(d)].除了对LiFePO4不同Li离子浓度与电压分布关系的计算,Zheng等[65]还对LiNi x Mn2-x O4/Li电池系统Ni掺杂下的电压分布进行了计算.作者首先构建了一个5 × 5 × 5的格子气模型,包含1000个可用位点.在模拟过程中,前500个MC模拟步作为弛豫过程,接下来的500个模拟步用于热力学量的计算,最终得到电压分布与x值之间的依赖关系,如图7(e)所示.可以看到随着x的增大,4.7 V电压平台的长度线性增大,4.1 V电压平台的长度线性减小,而总的电池容量则保持不变.当x = 0.5时,只观察到4.7 V的电压平台.有序到无序的转变和4.1、4.7 V两个电压平台的分裂都可以归因于锂离子与骨架结构之间的复杂相互作用.不同于上述对单个半电池的模拟,Kar等[19]通过对两个半电池(LiMn2O4正极和碳负极)进行建模,利用GCMC模拟给出了温度等参数变化对Li+嵌入与脱出开路电位、电池电流、电池电压、自由能等电池参数的影响.在模拟过程中,当固定温度T、体积V和化学势μ时,每个位置的能量可由式(5)给出 ...
1
... 在锂离子电池中,正极材料的离子输运特性直接决定了电池的容量与循环性能[62].LiFePO4[63]作为一种被广泛商用的正极材料,在平衡状态下,会自发地分裂出贫锂和富锂两相.如何快速获取相的分离过程十分重要.Xiao等[59]通过DFT计算得到KMC模拟的能垒输入,搭建了如图7(a)所示的模型.模拟结果显示,LiFePO4和FePO4之间形成有序的Li0.5FePO4相[图7(b)].Hin等[64]通过KMC模拟了Li x FePO4橄榄石纳米晶体中锂离子脱出与嵌入动力学特性.结果显示,锂离子嵌入过程中电池电压的变化包含以下几个阶段:①锂离子插入贫锂相导致电池电势下降;②富锂相Li1-β FePO4成核后,电势几乎恒定;③贫锂相完全演变为富锂相以后,电池的电势继续下降[图7(c)~(d)].除了对LiFePO4不同Li离子浓度与电压分布关系的计算,Zheng等[65]还对LiNi x Mn2-x O4/Li电池系统Ni掺杂下的电压分布进行了计算.作者首先构建了一个5 × 5 × 5的格子气模型,包含1000个可用位点.在模拟过程中,前500个MC模拟步作为弛豫过程,接下来的500个模拟步用于热力学量的计算,最终得到电压分布与x值之间的依赖关系,如图7(e)所示.可以看到随着x的增大,4.7 V电压平台的长度线性增大,4.1 V电压平台的长度线性减小,而总的电池容量则保持不变.当x = 0.5时,只观察到4.7 V的电压平台.有序到无序的转变和4.1、4.7 V两个电压平台的分裂都可以归因于锂离子与骨架结构之间的复杂相互作用.不同于上述对单个半电池的模拟,Kar等[19]通过对两个半电池(LiMn2O4正极和碳负极)进行建模,利用GCMC模拟给出了温度等参数变化对Li+嵌入与脱出开路电位、电池电流、电池电压、自由能等电池参数的影响.在模拟过程中,当固定温度T、体积V和化学势μ时,每个位置的能量可由式(5)给出 ...
2
... 在锂离子电池中,正极材料的离子输运特性直接决定了电池的容量与循环性能[62].LiFePO4[63]作为一种被广泛商用的正极材料,在平衡状态下,会自发地分裂出贫锂和富锂两相.如何快速获取相的分离过程十分重要.Xiao等[59]通过DFT计算得到KMC模拟的能垒输入,搭建了如图7(a)所示的模型.模拟结果显示,LiFePO4和FePO4之间形成有序的Li0.5FePO4相[图7(b)].Hin等[64]通过KMC模拟了Li x FePO4橄榄石纳米晶体中锂离子脱出与嵌入动力学特性.结果显示,锂离子嵌入过程中电池电压的变化包含以下几个阶段:①锂离子插入贫锂相导致电池电势下降;②富锂相Li1-β FePO4成核后,电势几乎恒定;③贫锂相完全演变为富锂相以后,电池的电势继续下降[图7(c)~(d)].除了对LiFePO4不同Li离子浓度与电压分布关系的计算,Zheng等[65]还对LiNi x Mn2-x O4/Li电池系统Ni掺杂下的电压分布进行了计算.作者首先构建了一个5 × 5 × 5的格子气模型,包含1000个可用位点.在模拟过程中,前500个MC模拟步作为弛豫过程,接下来的500个模拟步用于热力学量的计算,最终得到电压分布与x值之间的依赖关系,如图7(e)所示.可以看到随着x的增大,4.7 V电压平台的长度线性增大,4.1 V电压平台的长度线性减小,而总的电池容量则保持不变.当x = 0.5时,只观察到4.7 V的电压平台.有序到无序的转变和4.1、4.7 V两个电压平台的分裂都可以归因于锂离子与骨架结构之间的复杂相互作用.不同于上述对单个半电池的模拟,Kar等[19]通过对两个半电池(LiMn2O4正极和碳负极)进行建模,利用GCMC模拟给出了温度等参数变化对Li+嵌入与脱出开路电位、电池电流、电池电压、自由能等电池参数的影响.在模拟过程中,当固定温度T、体积V和化学势μ时,每个位置的能量可由式(5)给出 ...
<strong>MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li <i><sub>x</sub></i> FePO<sub>4</sub>, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis <i>a</i> without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO<sub>4</sub> and FePO<sub>4</sub> phases, and the Li<sub>0.5</sub>FePO<sub>4</sub> phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li <i><sub>x</sub></i> FePO<sub>4</sub> olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m<sup>-2</sup>. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi <i><sub>x</sub></i> Mn<sub>2</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> O<sub>4</sub> battery calculated by MC, <i>x</i> = 0.1, <i>x</i> = 0.2, <i>x</i> = 0.3, <i>x</i> = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn<sub>2</sub>O<sub>4</sub> anode and carbon cathode) and the Ni occupation of the cathode during discharge</strong>Fig. 7
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
2
... 在锂离子电池中,正极材料的离子输运特性直接决定了电池的容量与循环性能[62].LiFePO4[63]作为一种被广泛商用的正极材料,在平衡状态下,会自发地分裂出贫锂和富锂两相.如何快速获取相的分离过程十分重要.Xiao等[59]通过DFT计算得到KMC模拟的能垒输入,搭建了如图7(a)所示的模型.模拟结果显示,LiFePO4和FePO4之间形成有序的Li0.5FePO4相[图7(b)].Hin等[64]通过KMC模拟了Li x FePO4橄榄石纳米晶体中锂离子脱出与嵌入动力学特性.结果显示,锂离子嵌入过程中电池电压的变化包含以下几个阶段:①锂离子插入贫锂相导致电池电势下降;②富锂相Li1-β FePO4成核后,电势几乎恒定;③贫锂相完全演变为富锂相以后,电池的电势继续下降[图7(c)~(d)].除了对LiFePO4不同Li离子浓度与电压分布关系的计算,Zheng等[65]还对LiNi x Mn2-x O4/Li电池系统Ni掺杂下的电压分布进行了计算.作者首先构建了一个5 × 5 × 5的格子气模型,包含1000个可用位点.在模拟过程中,前500个MC模拟步作为弛豫过程,接下来的500个模拟步用于热力学量的计算,最终得到电压分布与x值之间的依赖关系,如图7(e)所示.可以看到随着x的增大,4.7 V电压平台的长度线性增大,4.1 V电压平台的长度线性减小,而总的电池容量则保持不变.当x = 0.5时,只观察到4.7 V的电压平台.有序到无序的转变和4.1、4.7 V两个电压平台的分裂都可以归因于锂离子与骨架结构之间的复杂相互作用.不同于上述对单个半电池的模拟,Kar等[19]通过对两个半电池(LiMn2O4正极和碳负极)进行建模,利用GCMC模拟给出了温度等参数变化对Li+嵌入与脱出开路电位、电池电流、电池电压、自由能等电池参数的影响.在模拟过程中,当固定温度T、体积V和化学势μ时,每个位置的能量可由式(5)给出 ...
<strong>MC simulation for phase transition and open circuit voltage of positive material: (a) MC simulation model (The above figure shows the distribution of Li ions in Li <i><sub>x</sub></i> FePO<sub>4</sub>, the corresponding energy diagram is shown below, the green balls represent Li ions); (b) the arrangement of Li ions along axis <i>a</i> without considering the interaction energy between charges. As shown in the figure, a clear phase boundary is formed between the LiFePO<sub>4</sub> and FePO<sub>4</sub> phases, and the Li<sub>0.5</sub>FePO<sub>4</sub> phase shrinks to a limited region; (c) MC simulation of constant current discharge process of Li <i><sub>x</sub></i> FePO<sub>4</sub> olivine nanocrystals at room temperature. The gray dots represent lithium atoms in the active particles with a surface current density of 0.5 A·m<sup>-2</sup>. The four groups are 0 s: initial solid solution, 0.0001 s: forming two Li-rich phases and combining together, 0.00056 s:Li-rich phase growth, 10.81 s: the Li-poor phase was almost completely consumed; (d) the relationship between the battery voltage and the concentration of Li ions in the active material; (e) the voltage distribution of Li/LiNi <i><sub>x</sub></i> Mn<sub>2</sub></strong><sub>-</sub><strong><i><sub>x</sub></i> O<sub>4</sub> battery calculated by MC, <i>x</i> = 0.1, <i>x</i> = 0.2, <i>x</i> = 0.3, <i>x</i> = 0.5, respectively; (f) the relationship between the open circuit potential of a full battery (LiMn<sub>2</sub>O<sub>4</sub> anode and carbon cathode) and the Ni occupation of the cathode during discharge</strong>Fig. 7
除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
2
... 除了对正极材料相变与开路电压的模拟,MC模拟同样适用于对石墨基、硅基负极材料相特征的模拟.Moon等[11]利用密度泛函理论(DFT)和KMC模拟,研究了锂离子插入c-Si和a-Si体系的热力学和动力学特征.DFT计算的形成能揭示了晶态硅与非晶硅在锂化过程中的相分离机制.晶态硅和非晶硅在锂化作用下的体积膨胀和相变趋势是相似的,而Li的扩散动力学在c-Si和a-Si之间则存在较大差异.在c-Si中,Li迁移势垒为0.6 eV,随着Li浓度的增加,迁移势垒迅速减小到至0.4 eV[图8(a)~(c)].为了利用KMC模拟非晶硅中锂的扩散,首先要利用体积函数推导出非晶硅中锂迁移势垒.通过KMC模拟发现锂在a-Si中的扩散系数比在c-Si中的扩散系数大一个数量级[图8(d)].这些研究有助于在原子尺度理解锂化作用的机理,进一步阐明硅化锂的相分离过程.此外,Persson等[66]也通过DFT、团簇扩展和MC模拟预测了Li x C6相图,预测结果在高Li浓度(x>0.5)与实验结果相吻合[图8(e)]. ...
... [66]Effect of Li ions arrangement on the transition barrier [(a)-(c)]; (d) diffusion coefficient of Li ion in a-Si and c-Si; (e) first-principles phase diagram obtained from Monte Carlo simulations. The phase regions are denoted in the following way: G (graphite), Ⅱ (stage Ⅱ), ⅡD (disordered stage Ⅱ), Ⅰ (stage Ⅰ), ⅠD (disordered stage Ⅰ), and Ⅱ′ (stage Ⅱ with 2×2 Li ordering)Fig. 8
... [67](a) schematic diagram of transition barrier between Cr ion and Li ions; (b) schematic diagram of lithium ions separated by Cr ions in each one-dimensional channel; (c) variation relation of the simulated capacity with the dopant amount and super-cell size. The numbers 200, 300, 500, 1000 and 2000 refer to the size of the super-cell being usedFig. 10
对于固态电解质,其离子输运的动力学特性更加受到关注,KMC在其中的应用更为广泛.由于典型的固态电解质存在三大特点[68]:①相邻位点之间的离子迁移容易发生;②可供迁移离子占据的位点的数量大于迁移离子的数量;③这些可用的位点连接成一个连续的扩散通路.这些特点使得格子气KMC可以很好地适用于固态电解质材料的模拟.Morgan等[20]根据石榴石构型固态电解质的结构特征[图11(a)~(c)],采用晶格气体MC模拟预测了石榴石结构固态电解质Li x La3Zr2O12的相关系数变化趋势,发现当xLi = 3(来自位置能量)和xLi = 6(来自最近邻斥力)时,其存在特别强的相关效应[图11(d)~(e)]. ...
1
... 对于固态电解质,其离子输运的动力学特性更加受到关注,KMC在其中的应用更为广泛.由于典型的固态电解质存在三大特点[68]:①相邻位点之间的离子迁移容易发生;②可供迁移离子占据的位点的数量大于迁移离子的数量;③这些可用的位点连接成一个连续的扩散通路.这些特点使得格子气KMC可以很好地适用于固态电解质材料的模拟.Morgan等[20]根据石榴石构型固态电解质的结构特征[图11(a)~(c)],采用晶格气体MC模拟预测了石榴石结构固态电解质Li x La3Zr2O12的相关系数变化趋势,发现当xLi = 3(来自位置能量)和xLi = 6(来自最近邻斥力)时,其存在特别强的相关效应[图11(d)~(e)]. ...
... [70];(h) 晶体尺寸与溶液pH值与氨含量依赖关系的相图.图中显示了在粒径分布为高斯分布的情况下,溶液的平均粒径及其对应的一阶标准差.这里显示的所有数据都是从计算模型中获取的.绿色区域表示次级颗粒较小,黄色区域表示次级颗粒较大.浅蓝点的二次粒径分布标准差较小,紫色点的二次粒径分布标准差较大.在pH值和氨含量方面,沉积较大(Dpart约8 μm)和较小(Dpart约4 μm)的二次活性颗粒的最佳操作条件也在图中突出显示(用红色圆圈表示)[71];(i) T = 1100 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(j) T = 1400 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(k) SEI膜的生成过程,一般分为吸附、解吸、表面扩散和钝化等步骤;(l)SEI膜厚度、充电速度与温度的依赖关系[73](a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (<i>D</i>part ~8 μm) and smaller (<i>D</i>part ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperatureFig.13
... [71];(i) T = 1100 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(j) T = 1400 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(k) SEI膜的生成过程,一般分为吸附、解吸、表面扩散和钝化等步骤;(l)SEI膜厚度、充电速度与温度的依赖关系[73](a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (<i>D</i>part ~8 μm) and smaller (<i>D</i>part ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperatureFig.13
... [72];(j) T = 1400 K下的不同Si/C比条件下,两种晶体在沉积速率 F = 0.1 ML/s下,6H-SiC在生长中的比例[72];(k) SEI膜的生成过程,一般分为吸附、解吸、表面扩散和钝化等步骤;(l)SEI膜厚度、充电速度与温度的依赖关系[73](a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (<i>D</i>part ~8 μm) and smaller (<i>D</i>part ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperatureFig.13
... [72];(k) SEI膜的生成过程,一般分为吸附、解吸、表面扩散和钝化等步骤;(l)SEI膜厚度、充电速度与温度的依赖关系[73](a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (<i>D</i>part ~8 μm) and smaller (<i>D</i>part ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperatureFig.13
... [73](a)-(g) the crystal growth process on the surface of a simple cubic lattice under different coordination; (h) Phase diagram of the dependence of crystal size on solution pH and ammonia content. The figure shows the mean particle size of the solution and its corresponding first standard deviation in the case of gaussian particle size distribution. All the data is taken from the computational model. Green areas indicate smaller secondary particles and yellow areas indicate larger secondary particles. The standard deviation of the secondary particle size distribution of the light blue dot is smaller, and the purple dot is larger. Optimum operating conditions, in terms of pH and ammonia content, for precipitating relatively larger (<i>D</i>part ~8 μm) and smaller (<i>D</i>part ~4 μm) sized secondary active particles, have also been highlighted within the figure (by red circles); (i) under different Si/C ratio conditions at 1100 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (j) under different Si/C ratio conditions at 1400 K, the ratio of 6H-SiC in the growth of the two crystals at the deposition rate <i>F</i> =0.1 ML/s; (k) the generation process of SEI film is generally divided into adsorption, desorption, surface diffusion and passivation, etc. (l) the dependence of SEI film thickness, charging speed and temperatureFig.13



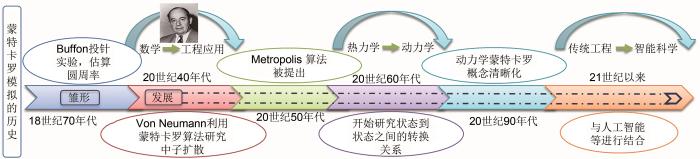
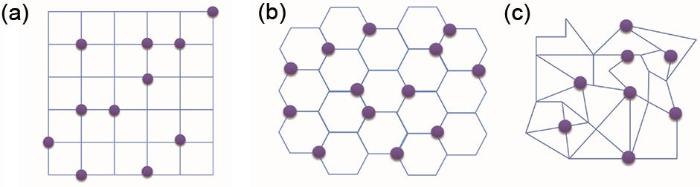
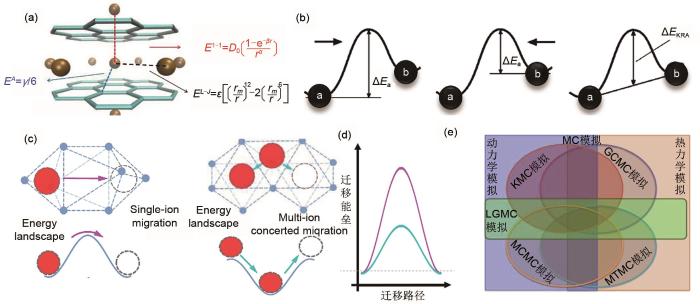
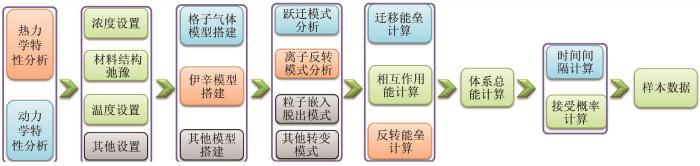
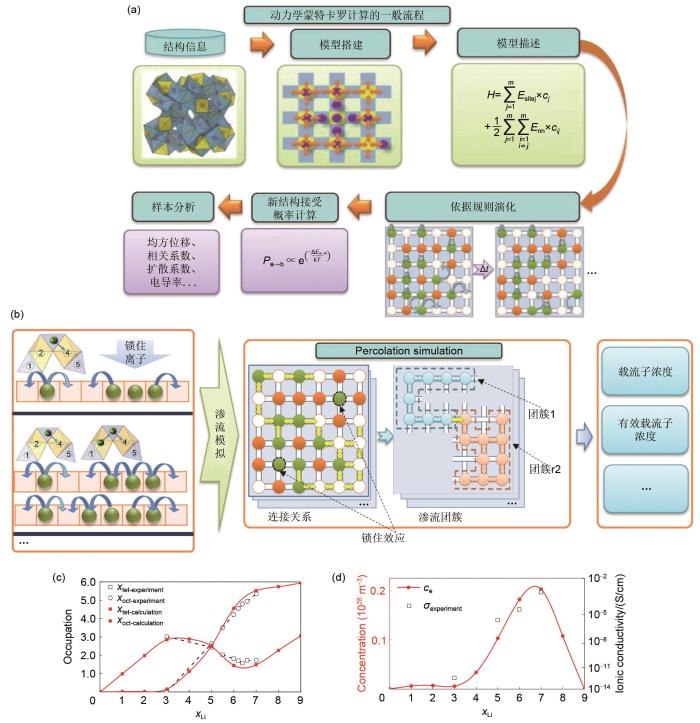
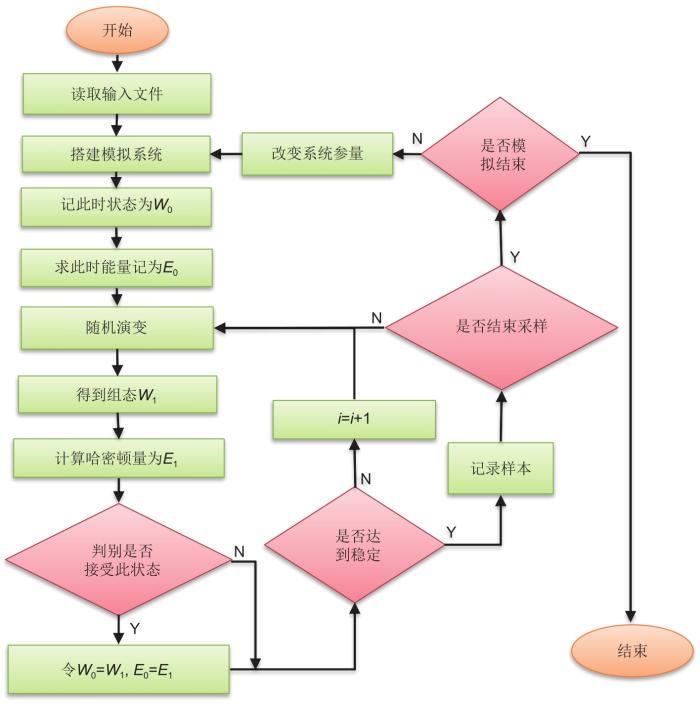
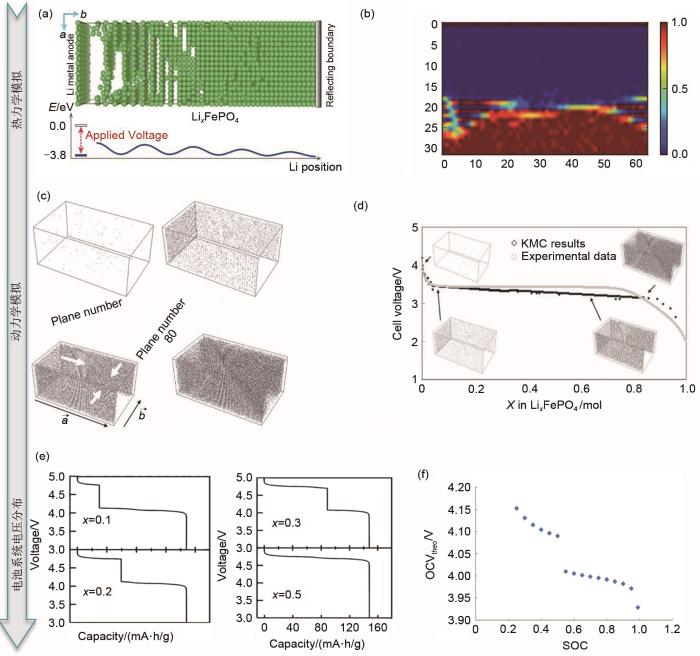
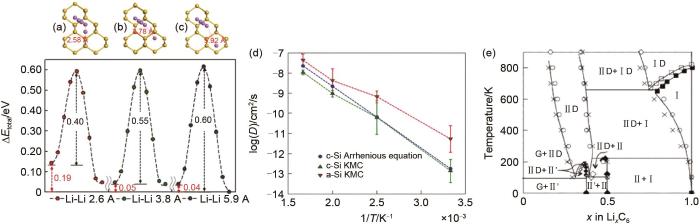
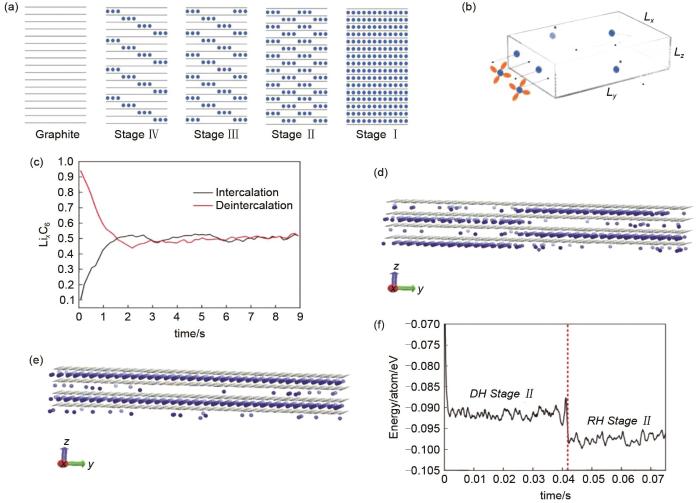
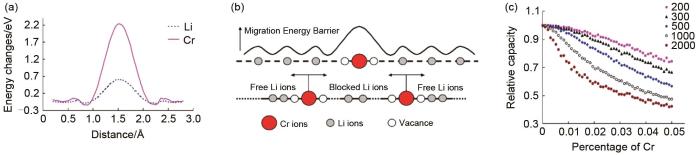
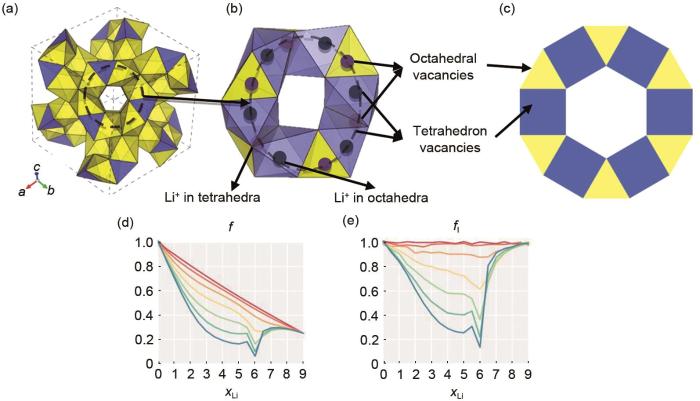
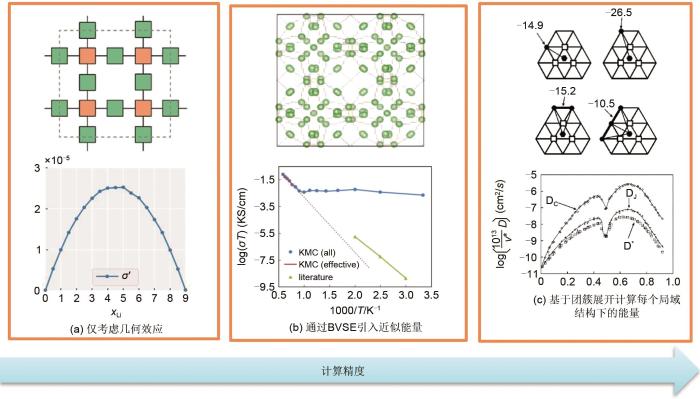
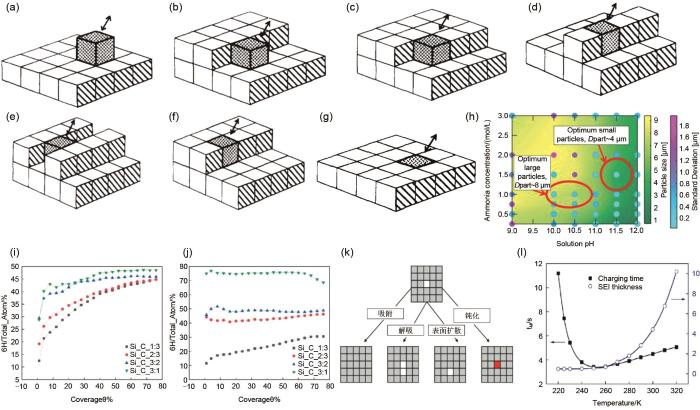

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}