In recent years, body-centered-cubic (bcc)-type anion framework has been considered as a deft structural design descriptor for identifying unexplored potential sulfide lithium-ion conductors (SICs). However, the bcc guidance must be completed, and other factors impacting lithium diffusion must be considered. Therefore, they are not always useful in the design of new lithium-ion conductors. In this study, based on previous works, we investigated the lithium-ion hopping and diffusion mechanisms in sulfides. We discussed how the local structural environment impacts lithium hopping from site to site in a micro-manner and how the chemical composition impacts diffusion in a macro-manner. In conclusion, we proved that the zero-transition-metal (0-TM) coordinated channel is essential for the facile hopping of lithium ions, and macro-diffusion can be expected in the materials only if the contents of 0-TM channels exceed the penetrating thresholds. Our study provides a more thorough interpretation of the sulfides' lithium diffusion mechanism and directly connects the structural and functional features of the material.
Keywords:lithium ionconductors
;
ion framework
;
lithium ion diffusion
;
diffusion coefficient
Fig. 1
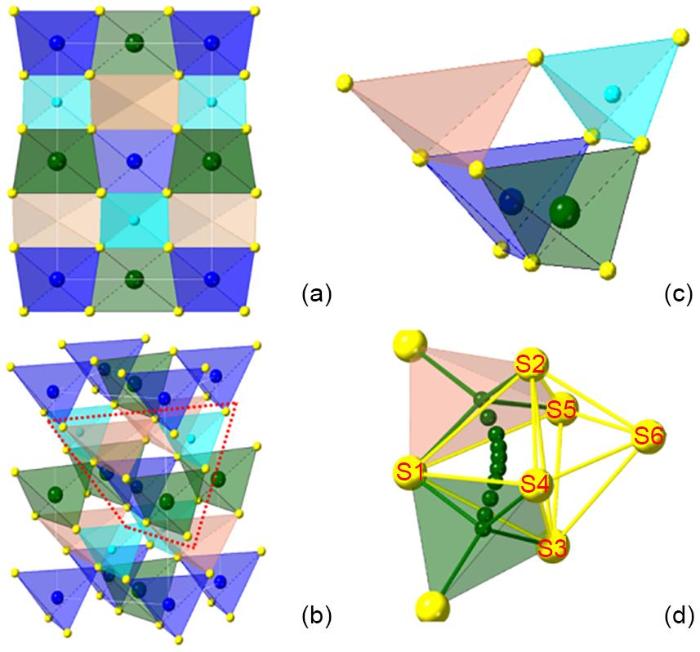
(a), (b) The unit cell of LZPS viewed from different directions. The LiS4, ZnS4, PS4 and interstitial site are colored by dark green, blue, cyan and pastel orange. The sulfur is labeled with yellow spheres. (c) The local environment involving the lithium hopping in the LZPS structure which is extracted from the red box in (b). (d) The diffusion path in the octahedral cage determined by NEB calculation
Fig. 2
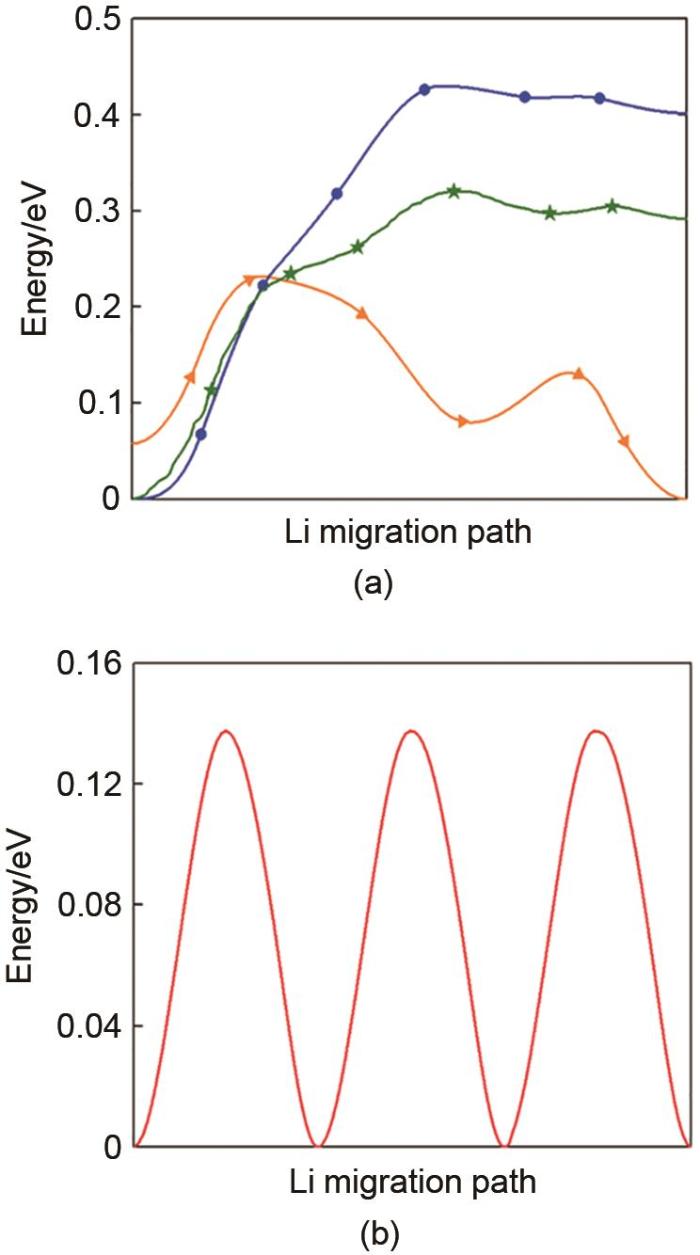
(a) The calculated energy landscape of diffusion path from the Li site to: the interstitial in the stoichiometry LZPS (blue color). The interstitial in a Li-Zn substitution local environment (green color). The Zn site in the same layer (orange color). (All the migration paths are scaled to the same length.) (b) The calculated energy landscape of diffusion path from the Li site to the interstitial site in a perfect bcc sulfur framework
Fig. 4
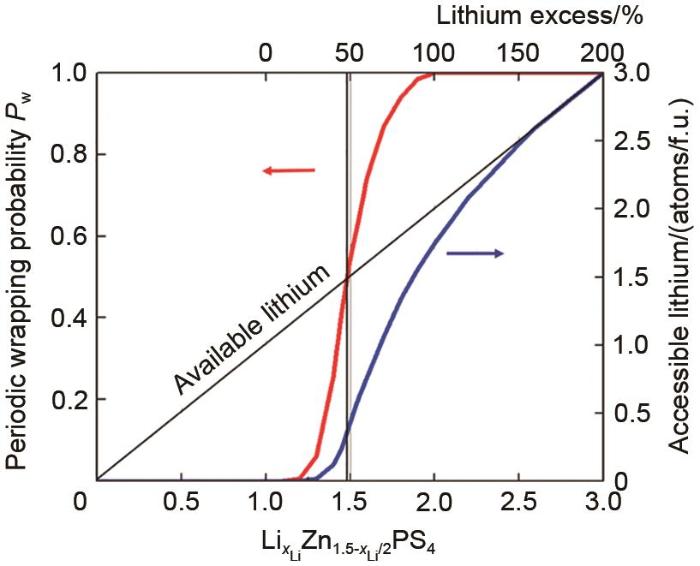
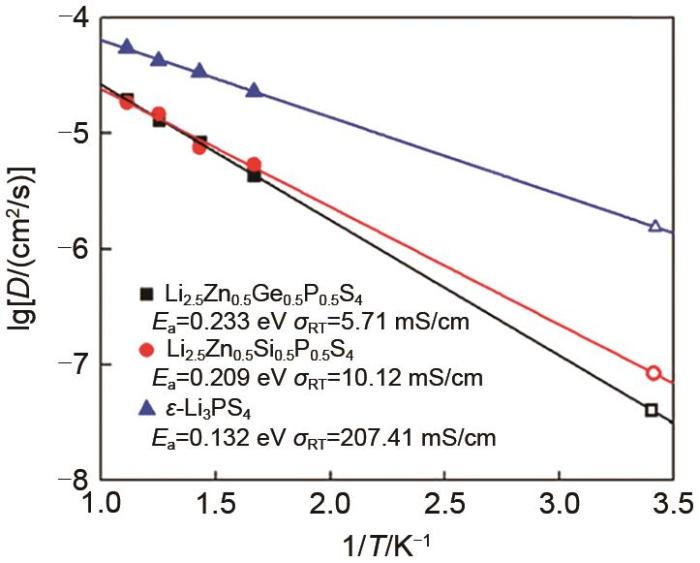
Periodic wrapping probabilities (red color line) and percolation thresholds (indicated by vertical bars). Accessible amount of lithium connecting to the percolating network (blue color line)
Fig. 5
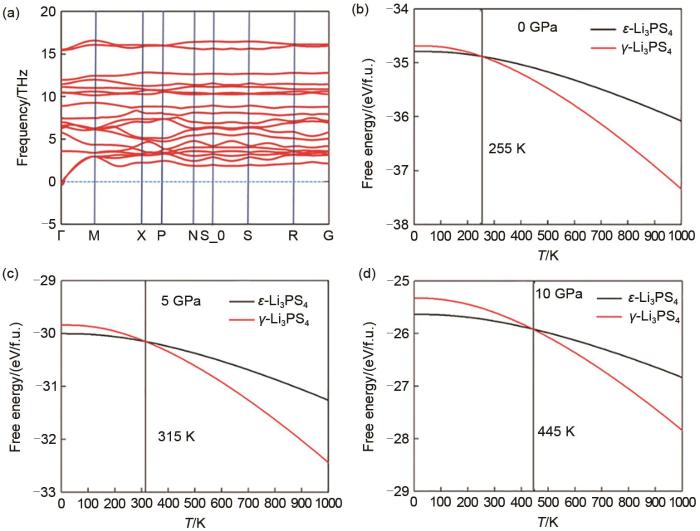
(a) The calculated phonon band structures for ε-Li3PS4; (b), (c), (d) The calculated Gibbs free energy of ε-Li3PS4 and γ-Li3PS4 at different pressures
Fig. 7
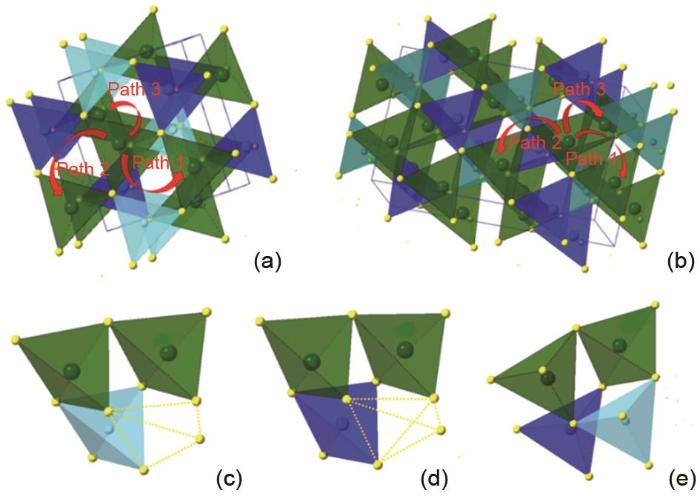
The unit cell of LZGS (a) and LZSS (b). The LiS4, ZnS4, GeS, and LiSi4 are colored by dark green, blue, pastel cyan and dark cyan. (c), (d), (e) The local environment involving the lithium hopping in the LZGS structure marked as path 1, path 2 and path 3 in (a)
Fig. 8
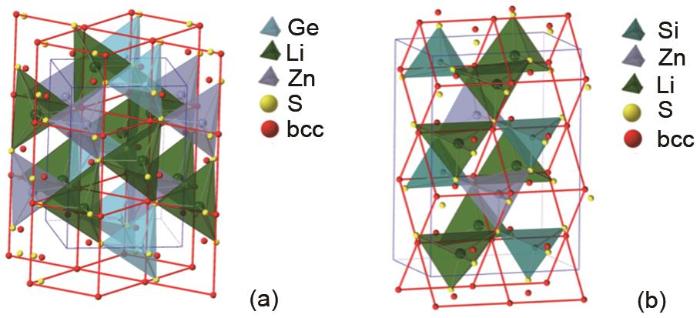
Ideal body-centered-tetragonal (bct) lattice (red) overlaid on the sulfur framework (yellow) of β-Li2ZnGeS4 (a) and Li2ZnSiS4 (b). The root-mean-square (rms) distances from the S atoms in the β-Li2ZnGeS4 and 0.56 Å in the Li2ZnSiS4 to the idealized bcc position are 0.53 Å and 0.56 Å respectively
Fig. 9
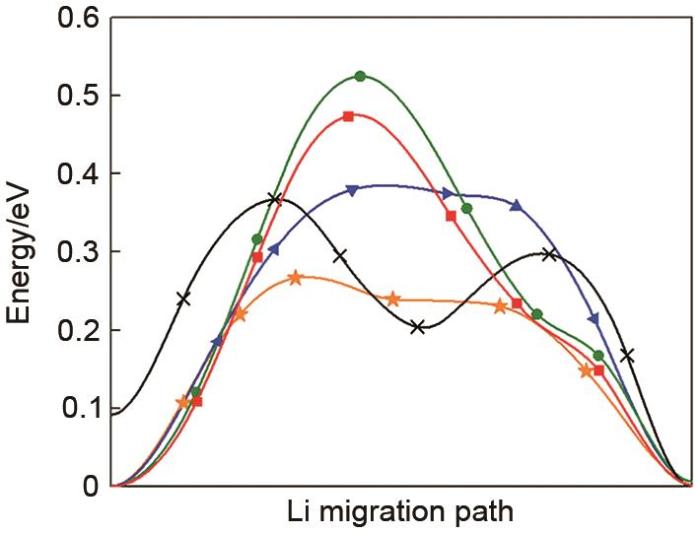
The calculated energy landscape of diffusion path in the LZGS from the Li site to the adjacent vacancy through the Oct(2LiS4-PS4) cage (orange color). The Oct(2LiS4-ZnS4) cage (blue color). The Tet(2LiS4-ZnS4-PS4) cage (green color). The Oct(2LiS4-VacS4) cage (black color). The Tet(2LiS4-VacS4-PS4) cage (red color)
Fig. 10
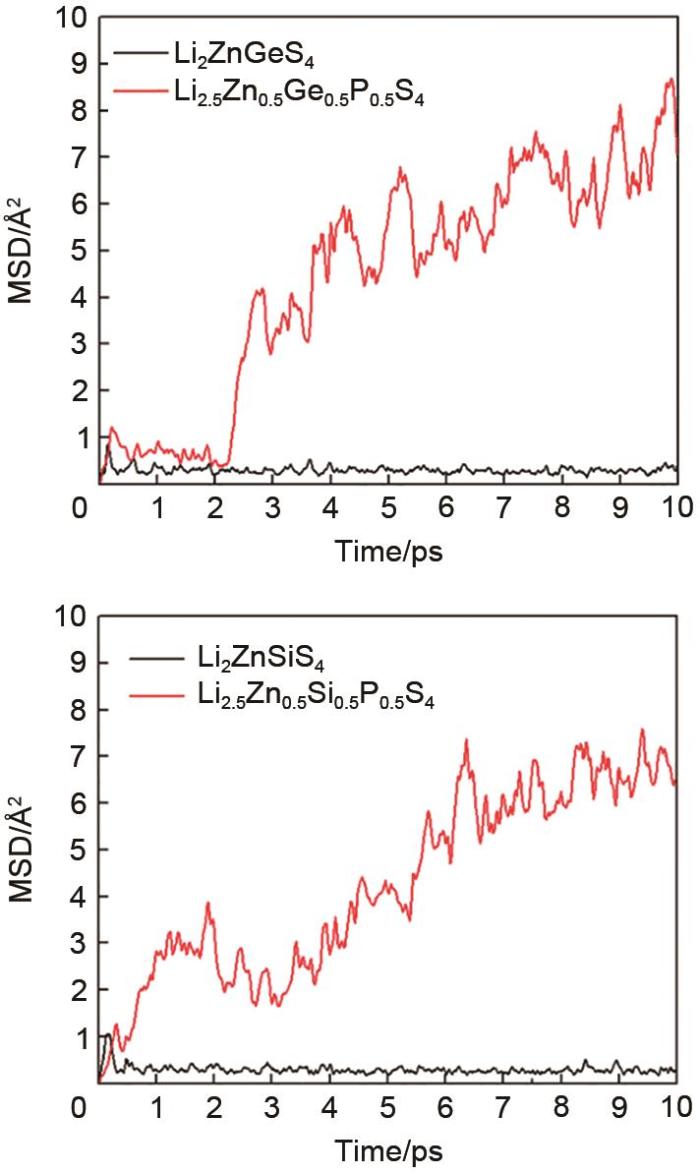
The mean square displacement (MSD) of lithium ions in the of β-Li2ZnGeS4 (left) and Li2ZnSiS4 (right) calculated from the AIMD simulation at 600 K. After the composition engineering, the diffusion is significantly enhanced, proving the success of our designing strategy
Fig. 11
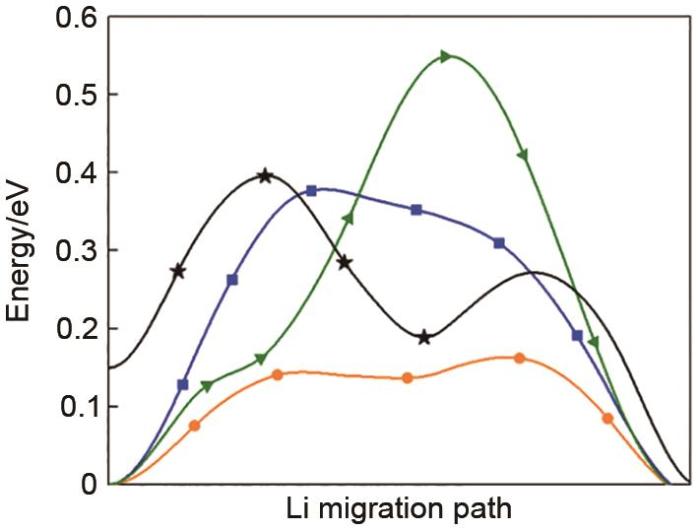
The calculated energy landscape of diffusion path in the LZSS from the Li site to the adjacent vacancy through the Oct(2LiS4-SiS4) cage (orange color). The Oct(2LiS4-ZnS4) cage (blue color). The Tet(2LiS4-ZnS4-PS4) cage (green color). The Oct(2LiS4-VacS4) cage (black color)
LIANG J N, LUO J, SUN Q, et al. Recent progress on solid-state hybrid electrolytes for solid-state lithium batteries[J]. Energy Storage Materials, 2019, 21: 308-334.
HE Y M, LU C Y, LIU S, et al. Interfacial incompatibility and internal stresses in all-solid-state lithium ion batteries[J]. Advanced Energy Materials, 2019, 9(36): doi: 10.1002/aenm.201901810.
LV F, WANG Z Y, SHI L Y, et al. Challenges and development of composite solid-state electrolytes for high-performance lithium ion batteries[J]. Journal of Power Sources, 2019, 441: 227175.
YU C, GANAPATHY S, VAN ECK E R H, et al. Accessing the bottleneck in all-solid state batteries, lithium-ion transport over the solid-electrolyte-electrode interface[J]. Nature Communications, 2017, 8(1): 1086.
WAN J, XIE J, MACKANIC D G, et al. Status, promises, and challenges of nanocomposite solid-state electrolytes for safe and high performance lithium batteries[J]. Materials Today Nano, 2018, 4: 1-16.
HAYASHI A, SAKUDA A, TATSUMISAGO M. Development of sulfide solid electrolytes and interface formation processes for bulk-type all-solid-state Li and Na batteries[J]. Frontiers in Energy Research, 2016, 4: 25.
LIU D, ZHU W, FENG Z, et al. Recent progress in sulfide-based solid electrolytes for Li-ion batteries[J]. Materials Science and Engineering: B, 2016, 213: 169-176.
YU C A, GANAPATHY S, DE KLERK N J J, et al. Unravelling Li-ion transport from picoseconds to seconds: Bulk versus interfaces in an argyrodite Li6PS5Cl-Li2S all-solid-state Li-ion battery[J]. Journal of the American Chemical Society, 2016, 138(35): 11192-11201.
KUHN A, GERBIG O, ZHU C B, et al. A new ultrafast superionic Li-conductor: Ion dynamics in Li11Si2PS12 and comparison with other tetragonal LGPS-type electrolytes[J]. Physical Chemistry Chemical Physics, 2014, 16(28): 14669-14674.
BRON P, JOHANSSON S, ZICK K, et al. Li10SnP2S12: An affordable lithium superionic conductor[J]. Journal of the American Chemical Society, 2013, 135(42): 15694-15697.
SEINO Y, OTA T, TAKADA K, et al. A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries[J]. Energy & Environmental Science, 2014, 7(2): 627-631.
Clement R J, Lun Z, Ceder G. Cation-disordered rocksalt transition metal oxides and oxyfluorides for high energy lithium-ion cathodes[J]. Energy & environmental science. 2020, 13(2): 345-373.
HE X F, BAI Q, LIU Y S, et al. Crystal structural framework of lithium super-ionic conductors[J]. Advanced Energy Materials, 2019, 9(43): doi: 10.1002/aenm.201902078.
RICHARDS W D, WANG Y, MIARA L J, et al. Design of Li1+2xZn1-xPS4, a new lithium ion conductor[J]. Energy & Environmental Science, 2016, 9(10): 3272-3278.
SUZUKI N, RICHARDS W D, WANG Y, et al. Synthesis and electrochemical properties of I-4-type Li1+2xZn1-xPS4 solid electrolyte[J]. Chemistry of Materials, 2018, 30(7): 2236-2244.
KAUP K, LALÈRE F, HUQ A, et al. Correlation of structure and fast ion conductivity in the solid solution series Li1+2xZn1-xPS4[J]. Chemistry of Materials, 2018, 30(3): 592-596.
SHYUE P O, WILLIAM D R, ANUBHAV J, et al. Python materials genomics (pymatgen): A robust, open-source python library for materials analysis[J]. Computational Materials Science, 2013, 68: 314-319.
KRESSE G, HAFNER J. Ab initio molecular dynamics for liquid metals[J]. Physical Review B, 1993, 47(1): 558-561.
KRESSE G, FURTHMÜLLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169-11186.
HENKELMAN G, UBERUAGA B P, JóNSSON H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths[J]. The Journal of Chemical Physics, 2000, 113(22): 9901.
HENKELMAN G, JÓNSSON H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points[J]. The Journal of Chemical Physics, 2000, 113(22): 9978-9985.
TOGO A, CHAPUT L, TANAKA I, et al. First-principles phonon calculations of thermal expansion in Ti3SiC2, Ti3AlC2, and Ti3GeC2[J]. Physical Review B, 2010, 81(17): 174301.
VERLET L. Computer "experiments" on classical fluids. I. thermodynamical properties of lennard-jones molecules[J]. Physical Review, 1967, 159(1): 98-103.
MEHRER, H. Diffusion in solids: Fundamentals, methods, materials, diffusion-controlled processes[J]. Springer series in solid state science, 2007, 155.
URBAN A, LEE J, CEDER G. The configurational space of rocksalt-type oxides for high-capacity lithium battery electrodes[J]. Advanced Energy Materials, 2014, 4(13): 1400478.
LEE J, URBAN A, LI X, et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries[J]. Science, 2014, 343(6170): 519-522.
JI H W, URBAN A, KITCHAEV D A, et al. Hidden structural and chemical order controls lithium transport in cation-disordered oxides for rechargeable batteries[J]. Nature Communications, 2019, 10: 592.
URBAN A, MATTS I, ABDELLAHI A, et al. Computational design and preparation of cation-disordered oxides for high-energy-density Li-ion batteries[J]. Advanced Energy Materials, 2016, 6(15): 1600488.
HUANG Y, LIU L, ZHU Y Y, et al. A new model on cation distribution in cation-disordered Li1+xTM1-xO2 cathodes[J]. Solid State Ionics, 2020, 351: 115341.
HOMMA K, YONEMURA M, KOBAYASHI T, et al. Crystal structure and phase transitions of the lithium ionic conductor Li3PS4[J]. Solid State Ionics, 2010, 182(1): 53-58.
KIM J S, JUNG W D, CHOI S J, et al. Thermally induced S‑sublattice transition of Li3PS4 for fast lithium-ion conduction[J]. J. Phys. Chem. Lett. 2018, 9, 5592-5597.
YANG J J, TSE J S. First-principles molecular simulations of Li diffusion in solid electrolytes Li3PS4[J]. Computational Materials Science, 2015, 107: 134-138.
LIM M S, JHI S H. First-principles study of lithium-ion diffusion in β-Li3PS4 for solid-state electrolytes[J]. Current Applied Physics, 2018, 18(5): 541-545.
HUANG Y, WU K, CHENG J N, et al. Li2ZnGeS4: A promising diamond-like infrared nonlinear optical material with high laser damage threshold and outstanding second-harmonic generation response[J]. Dalton Transactions, 2019, 48(14): 4484-4488.
LI G M, CHU Y, ZHOU Z X. From AgGaS2 to Li2ZnSiS4: Realizing impressive high laser damage threshold together with large second-harmonic generation response[J]. Chemistry of Materials, 2018, 30(3): 602-606.
ZHOU L D, ASSOUD A, SHYAMSUNDER A, et al. An entropically stabilized fast-ion conductor: Li3.25[Si0.25P0.75]S4[J]. Chemistry of Materials, 2019, 31(19): 7801-7811.
HARM S, HATZ A K, MOUDRAKOVSKI I, et al. Lesson learned from NMR: Characterization and ionic conductivity of LGPS-like Li7SiPS8[J]. Chemistry of Materials, 2019, 31(4): 1280-1288.
KANNO R, MURAYAMA M. Lithium ionic conductor thio-LISICON the Li2S-GeS2-P2S5 system[J]. Journal of the Electrochemical Society, 2001, 148(7): A742-A746.
LEKSE J W, LEVERETT B M, LAKE C H, et al. Synthesis, physicochemical characterization and crystallographic twinning of Li2ZnSnS4[J]. Journal of Solid State Chemistry, 2008, 181(12): 3217-3222.
ZHANG J H, CLARK D J, BRANT J A, et al. α -Li2ZnGeS4: A wide-bandgap diamond-like semiconductor with excellent balance between laser-induced damage threshold and second harmonic generation response[J]. Chemistry of Materials, 2020, 32(20): 8947-8955.
LI G M, CHU Y, LI J, et al. Li2CdSiS4, a promising IR NLO material with a balanced Eg and SHG response originating from the effect of Cd with d10 configuration[J]. Dalton Transactions, 2020, 49(6): 1975-1980.
LI Y L, FAN W L, SUN H G, et al. Electronic, optical and lattice dynamic properties of the novel diamond-like semiconductors Li2CdGeS4 and Li2CdSnS4[J]. Journal of Physics: Condensed Matter, 2011, 23(22): 225401.
BRANT J A, CLARK D J, KIM Y S, et al. Outstanding laser damage threshold in Li2MnGeS4 and tunable optical nonlinearity in diamond-like semiconductors[J]. Inorganic Chemistry, 2015, 54(6): 2809-2819.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}