储能科学与技术 ›› 2024, Vol. 13 ›› Issue (9): 2884-2906.doi: 10.19799/j.cnki.2095-4239.2024.0699
邓斌1( ), 华海明1, 张与之1, 王晓旭1(), 张林峰1,2
), 华海明1, 张与之1, 王晓旭1(), 张林峰1,2
收稿日期:2024-07-29
修回日期:2024-08-26
出版日期:2024-09-28
发布日期:2024-09-20
通讯作者:
王晓旭
E-mail:dengb@dp.tech;wangxx@dp.tech
作者简介:邓斌(1992—),男,硕士,主要从事电池材料的仿真开发,E-mail: dengb@dp.tech;
Bin DENG1(), Haiming HUA1, Yuzhi ZHANG1, Xiaoxu WANG1(), Linfeng ZHANG1,2
Received:2024-07-29
Revised:2024-08-26
Online:2024-09-28
Published:2024-09-20
Contact:
Xiaoxu WANG
E-mail:dengb@dp.tech;wangxx@dp.tech
摘要:
深度势能模型(deep potential,DP)通过先进的机器学习技术,从大量的原子结构和能量数据中提取知识,构建出高精度的势能面。这一创新方法有效突破了传统力场方法的局限,为材料科学领域带来了新的视角。本文概述了DP模型和软件的基本原理、开发进展与应用流程,回顾了其在电化学储能材料设计中的应用,展示了DP模型在揭示电池材料微观结构和动力学行为方面的优势。在正负极材料的研究中,精确描述脱嵌锂过程中材料的结构演变和自由能变化;在固态电解质的研究中,精确描述了材料结构与离子输运行为;在电解液的研究中,不仅提高了对溶液动态结构和性质的认识,还为氧化还原电位、酸度等物理化学性质的精确计算提供了新策略;在界面的研究中,准确解析了界面形成过程中的结构演变以及性质。这些对材料的准确描述均有利于加速对能源材料的开发。同时,指出了DP模型在电池材料模拟中仍需改进的问题,并展望了其在电池材料设计和优化中的潜在应用前景。结果说明,深度势能模型作为一种强大的计算工具,在电化学储能材料的研究中展现出巨大的应用潜力。通过不断的模型优化和算法创新,DP模型有望在未来的材料设计和电池技术发展中发挥更加关键的作用。
中图分类号:
邓斌, 华海明, 张与之, 王晓旭, 张林峰. 深度势能方法及其在电化学储能材料中的应用[J]. 储能科学与技术, 2024, 13(9): 2884-2906.
Bin DENG, Haiming HUA, Yuzhi ZHANG, Xiaoxu WANG, Linfeng ZHANG. Deep potential model: Applications and insights for electrochemical energy storage materials[J]. Energy Storage Science and Technology, 2024, 13(9): 2884-2906.
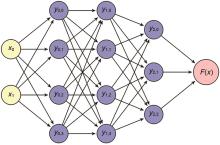
图1
多层神经网络"
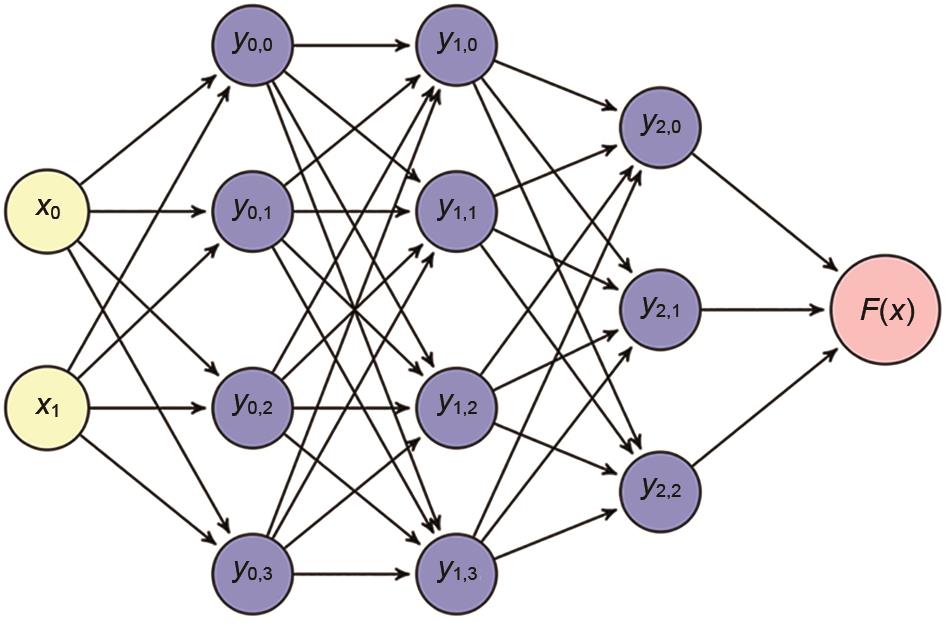

图2
下游任务样本效率对比分析。横坐标表示所需的下游数据量,纵坐标表示RMSE能量或力预测的收敛性[5]"
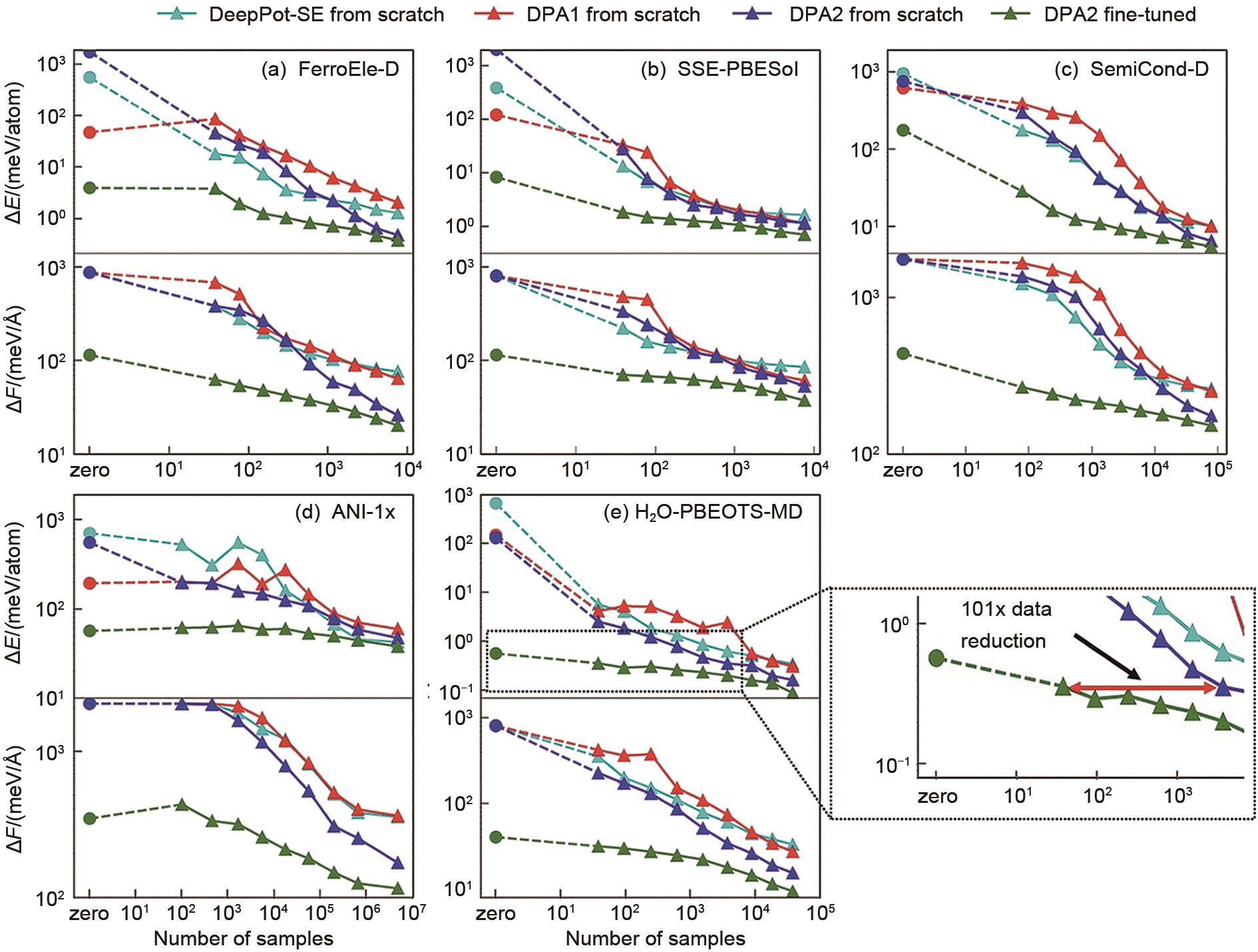
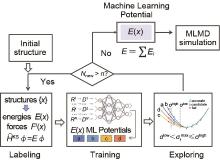
图3
DP-GEN自动化流程[21]"
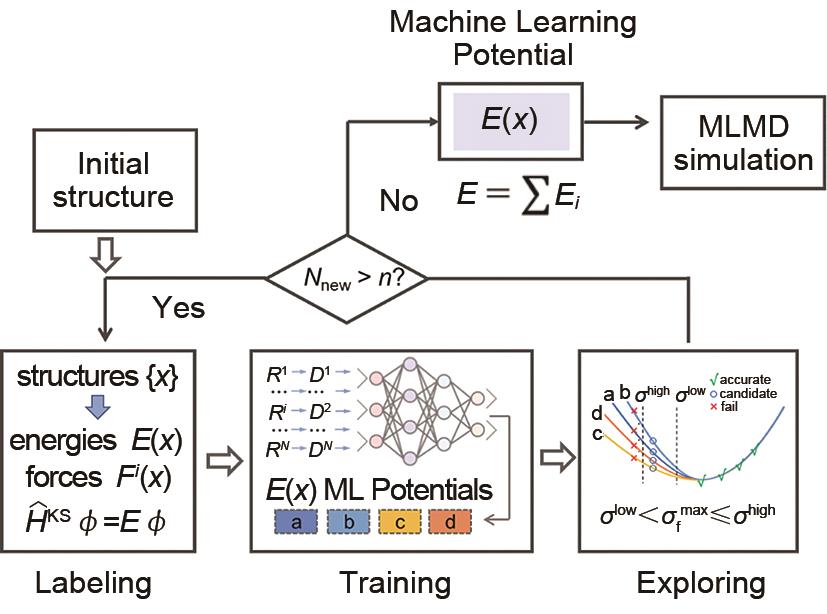
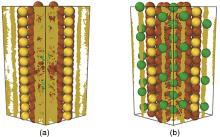
图4
(a) 低Na和 (b) 高Na含量C8S材料中Na+ 的扩散轨迹[23]"

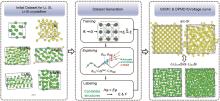
图5
Li-S-Si体系DP模型训练及检验过程[25]"
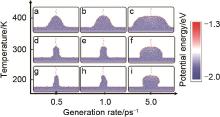
图6
不同温度(T)和生成速率(Rg)下不均匀沉积快照。 Rg=0.5 ps-1 (a, d, g),1.0 ps-1 (b, e, h) 和5.0 ps-1 (c, f, i); T=400 K (a~c), 300 K (d~f), 200 K, (g~i)[26]"
图7
基于增强采样DPMD模拟的CoO2 模型产氧反应路径图[31]"
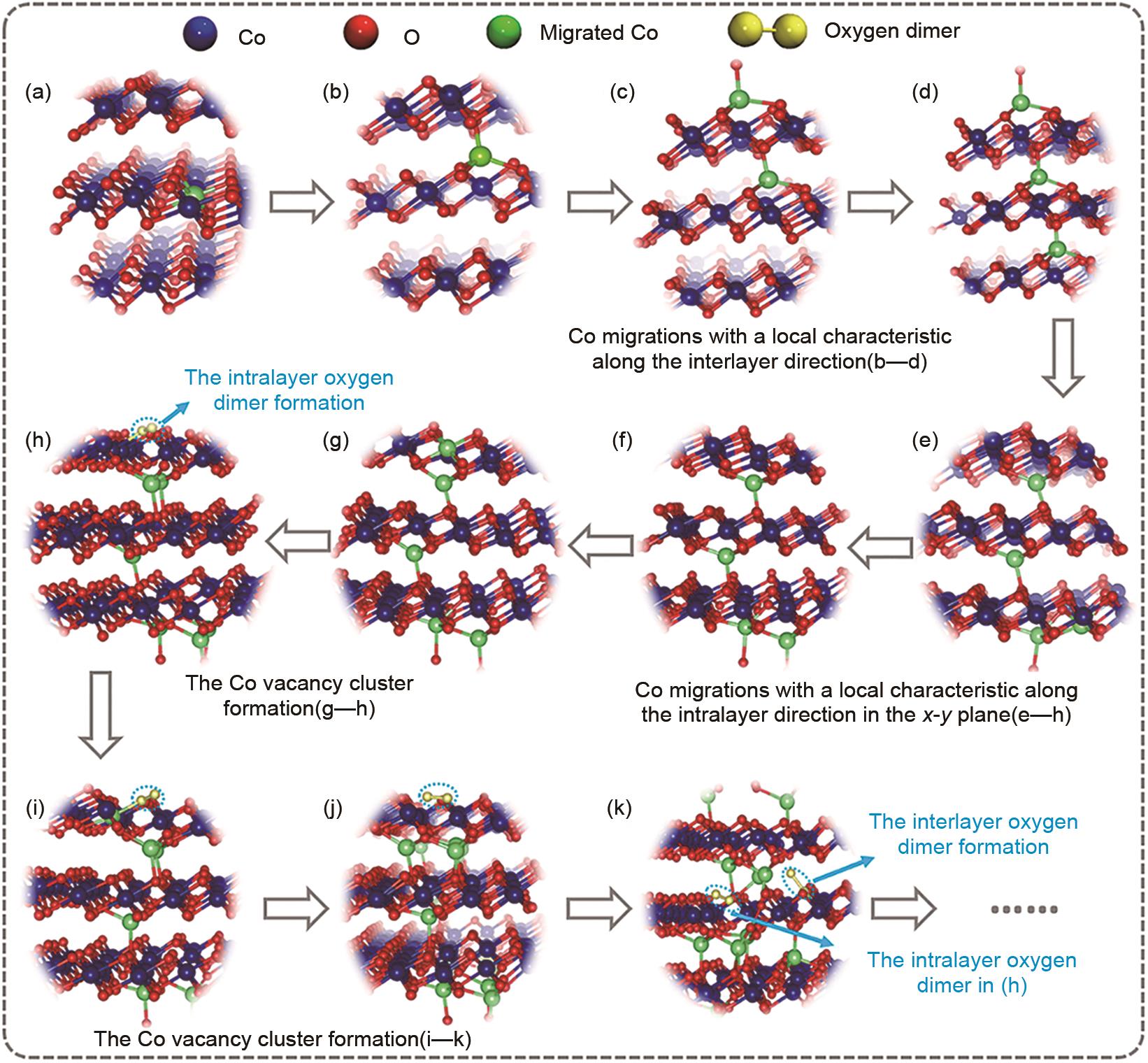
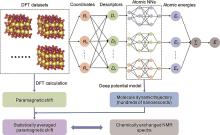
图8
基于DPMD的固态核磁化学位移计算流程[33]"

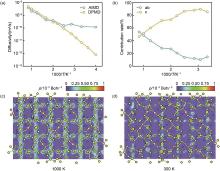
图9
Li10GeP2S12 晶格中的Li+ 扩散行为。(a) 用AIMD和DPMD模拟的扩散系数Arrhenius图。(b) 锂离子扩散的尺寸贡献随温度倒数变化。(c) 1000 K和 (d) 300 K时Li10GeP2S12 中的锂离子概率密度填色图[37]"
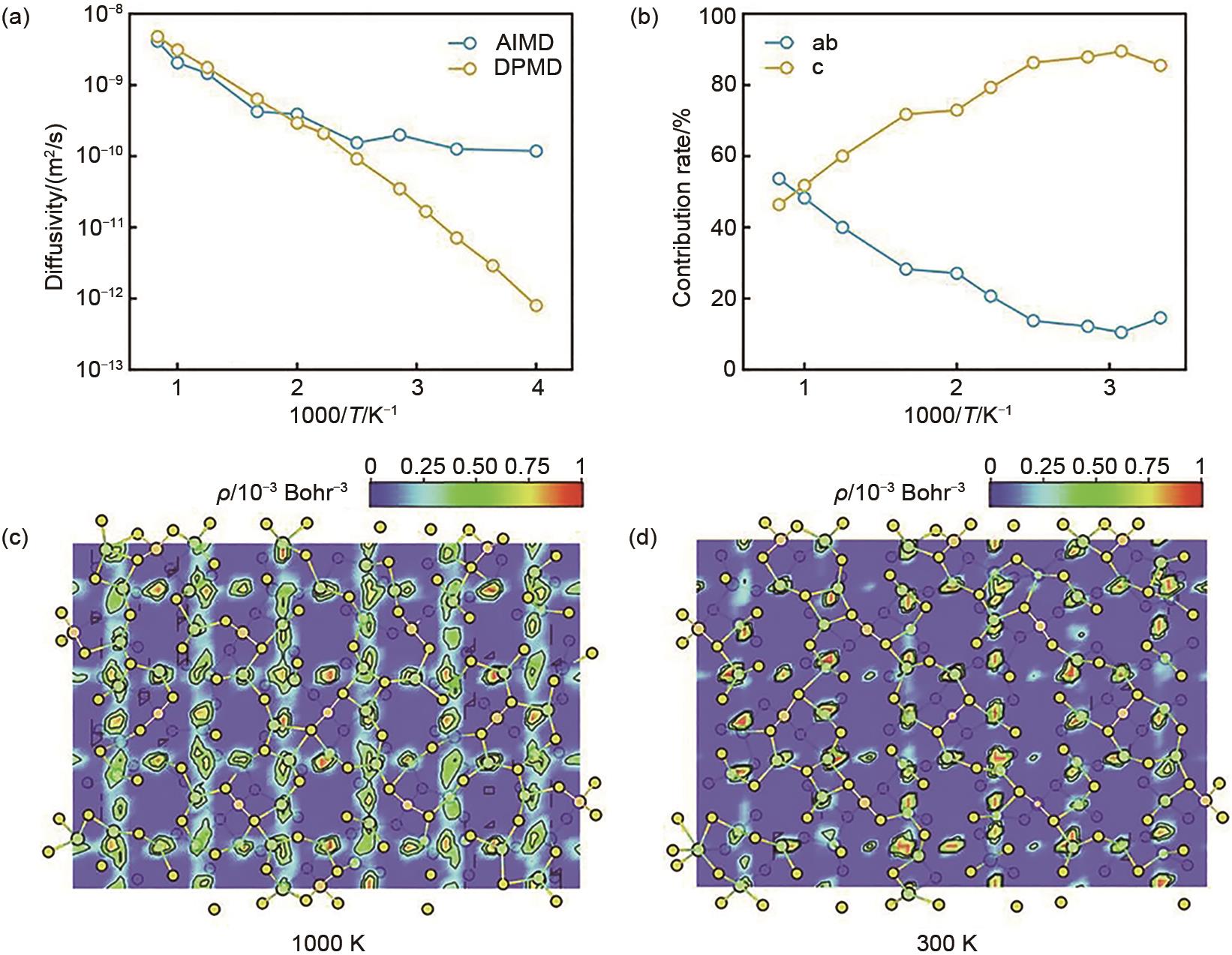

图10
电导率 (σ) 和Haven比的温度依赖性 (a, b, c) DPMD的离子电导率Arrhenius图;(d, e, f) Haven比随温度倒数变化的规律[39]"
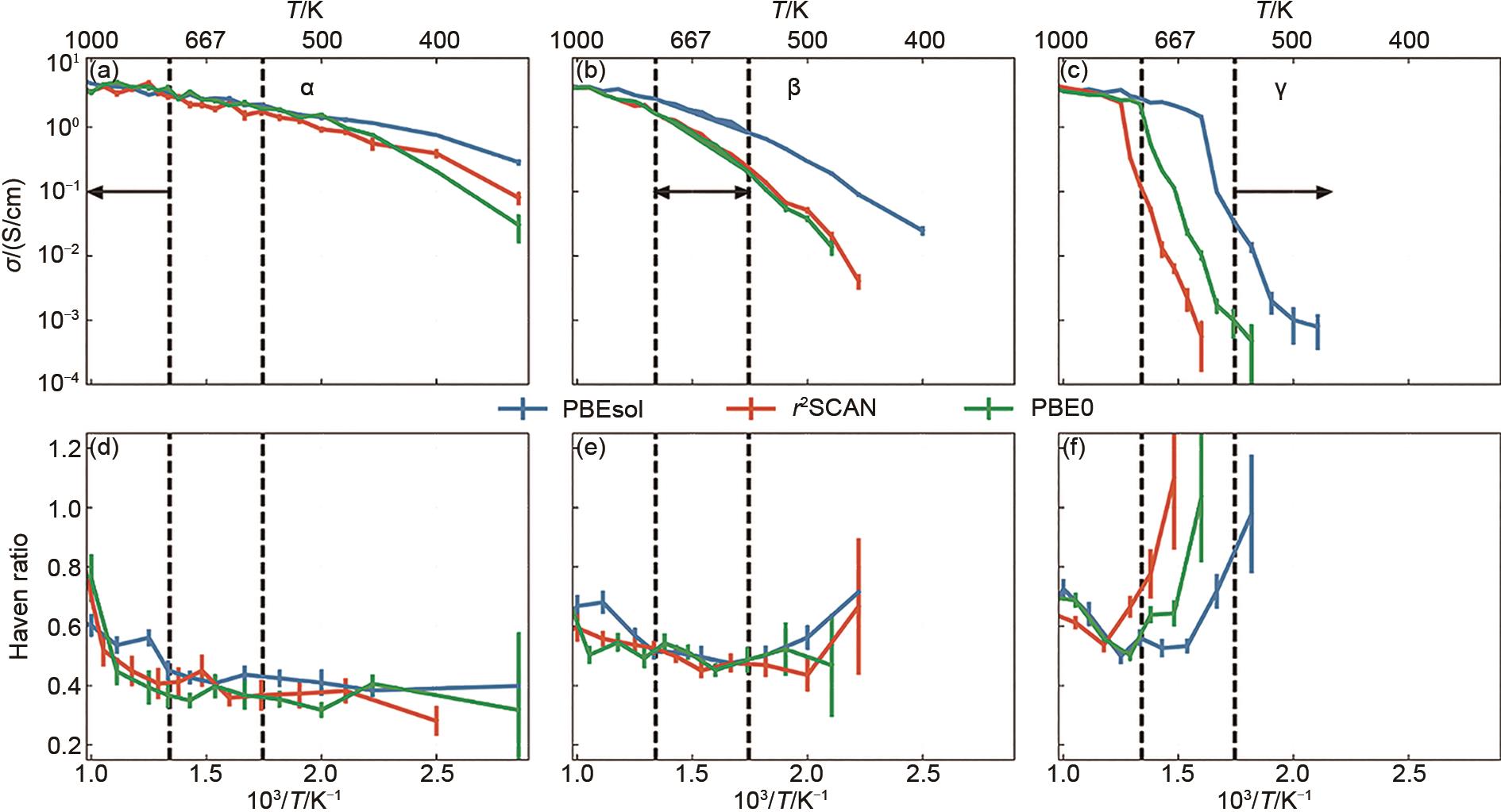

图11
沿Li3TiCl6 (a) [010] 和 (b) [110]视角方向的层内锂离子运动轨迹;(c) 对应 (a) 和 (b) 的Li原子位移随时间演化;沿Li3TiCl6 (d) [010]和 (e) [110]视角方向向的层间锂离子运动轨迹示意图;(f) 对应 (d) 和 (e) 的Li原子的位移随时间演化[44]"
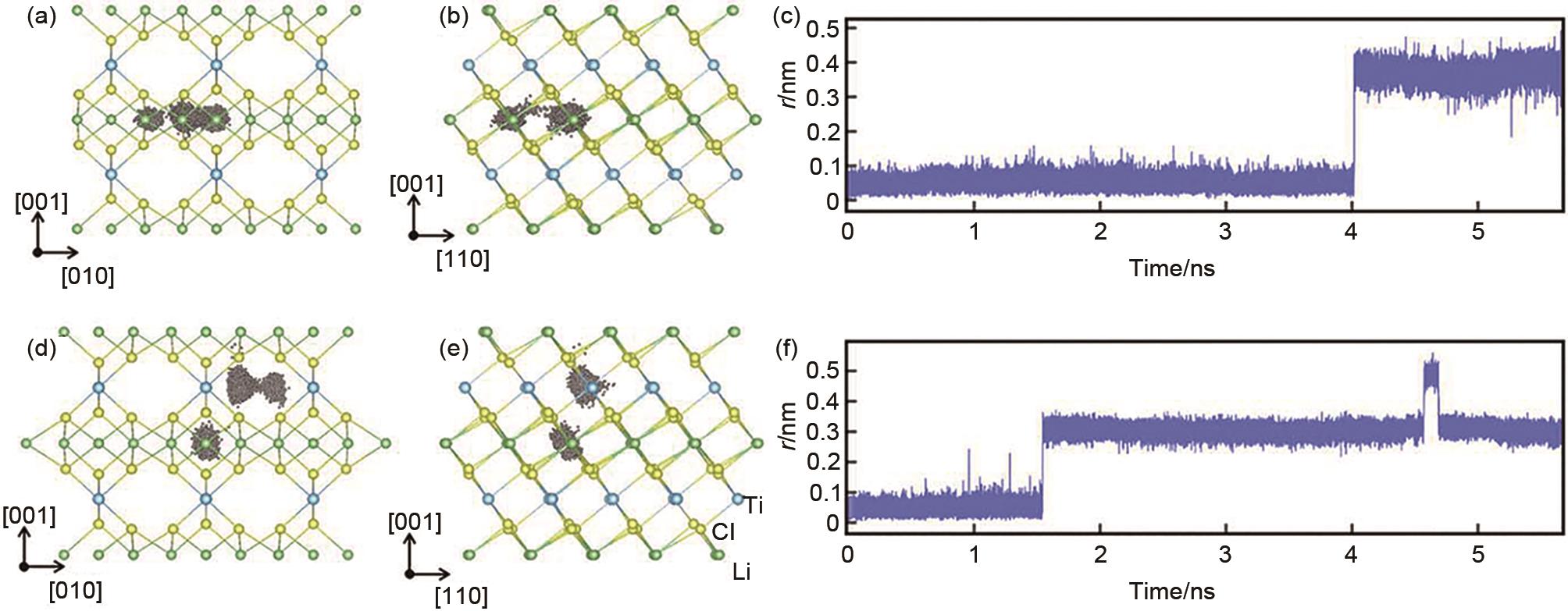
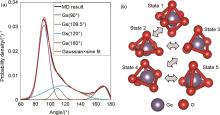
图12
(a) O-Ge-O角分布(分解为高斯函数和正弦函数的乘积);(b) 液态/玻璃态Ge原子氧环境的可能转变:1—八面体,2—方形金字塔,3—三角双金字塔,4—过渡结构,5—四面体[49]"
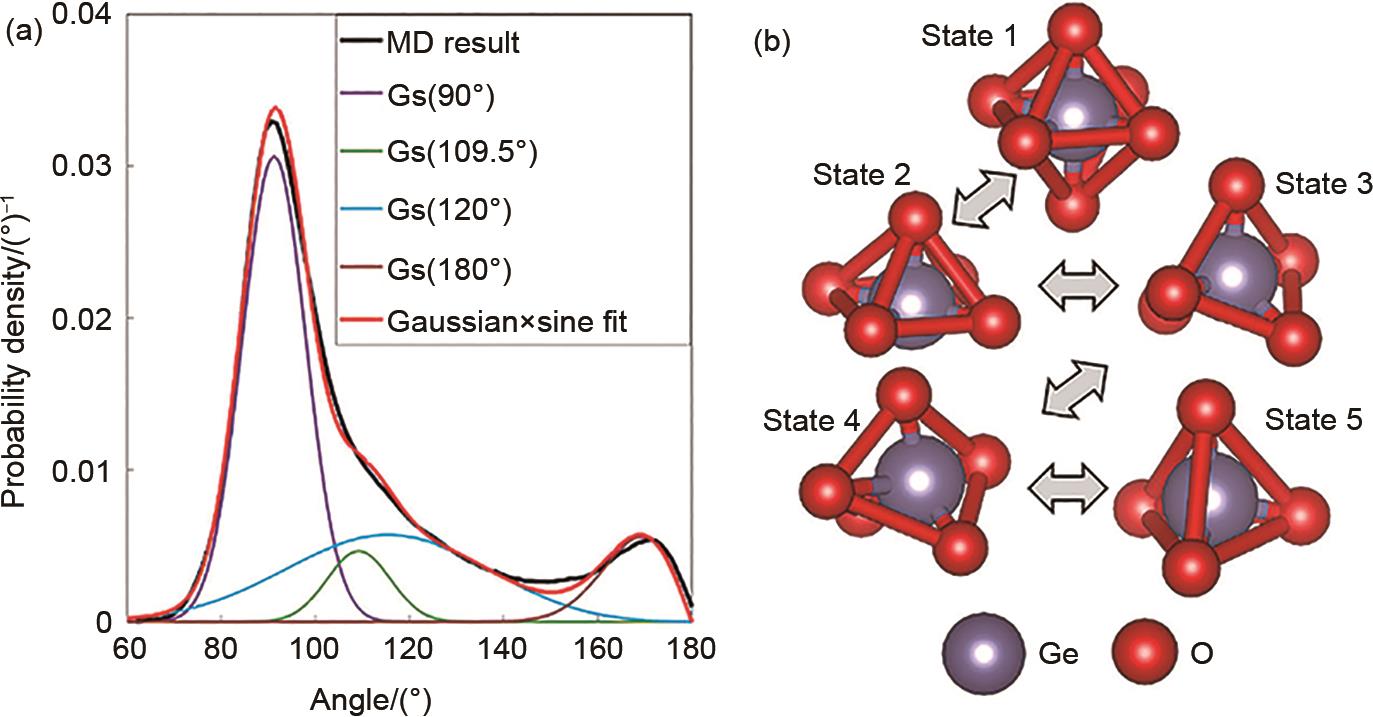

图13
非晶Li-La-Zr-O系统深度势能预训练流程[53]"
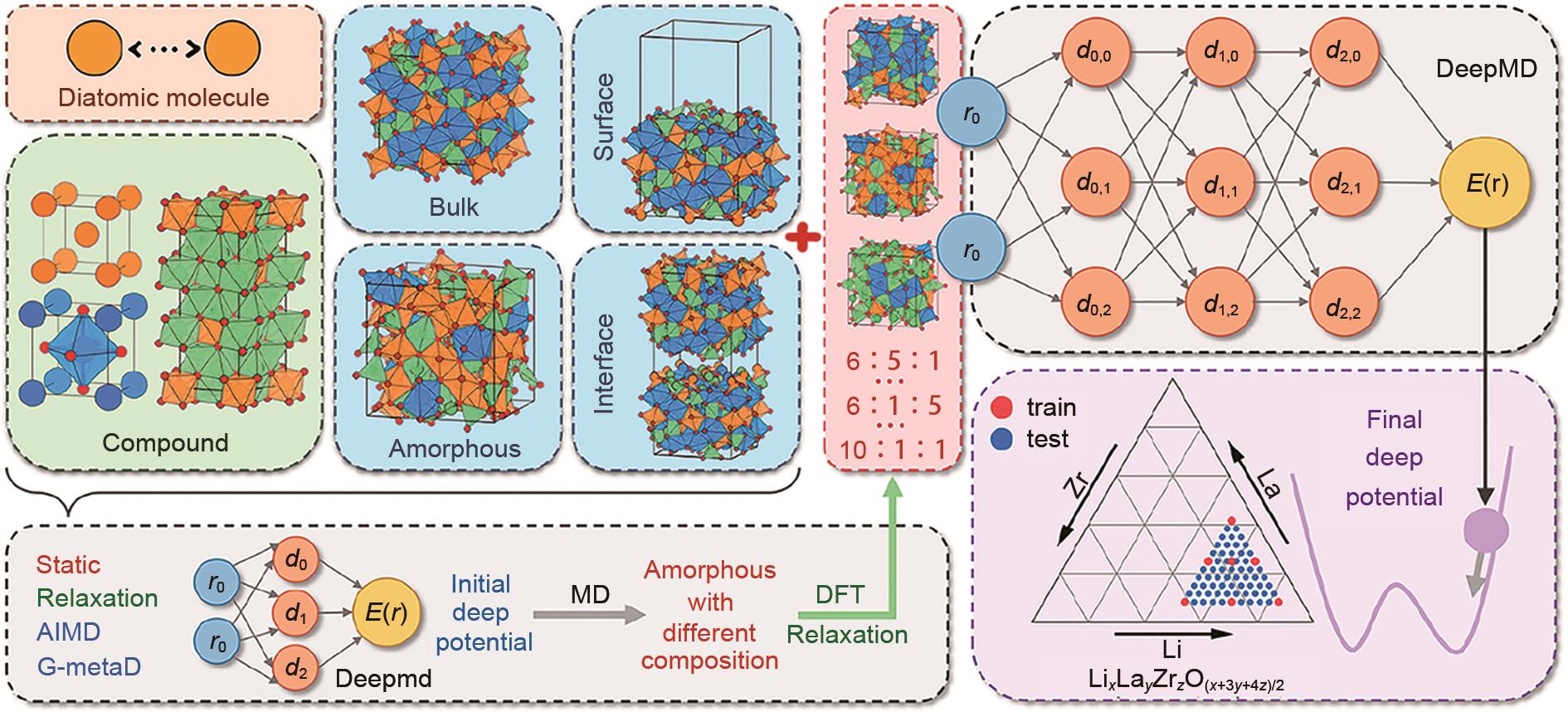

图14
酸度与氧化还原电位自动化工作流 (a) 初始化;(b) DP势函数训练过程;(c) DPMD自由能计算[11]"
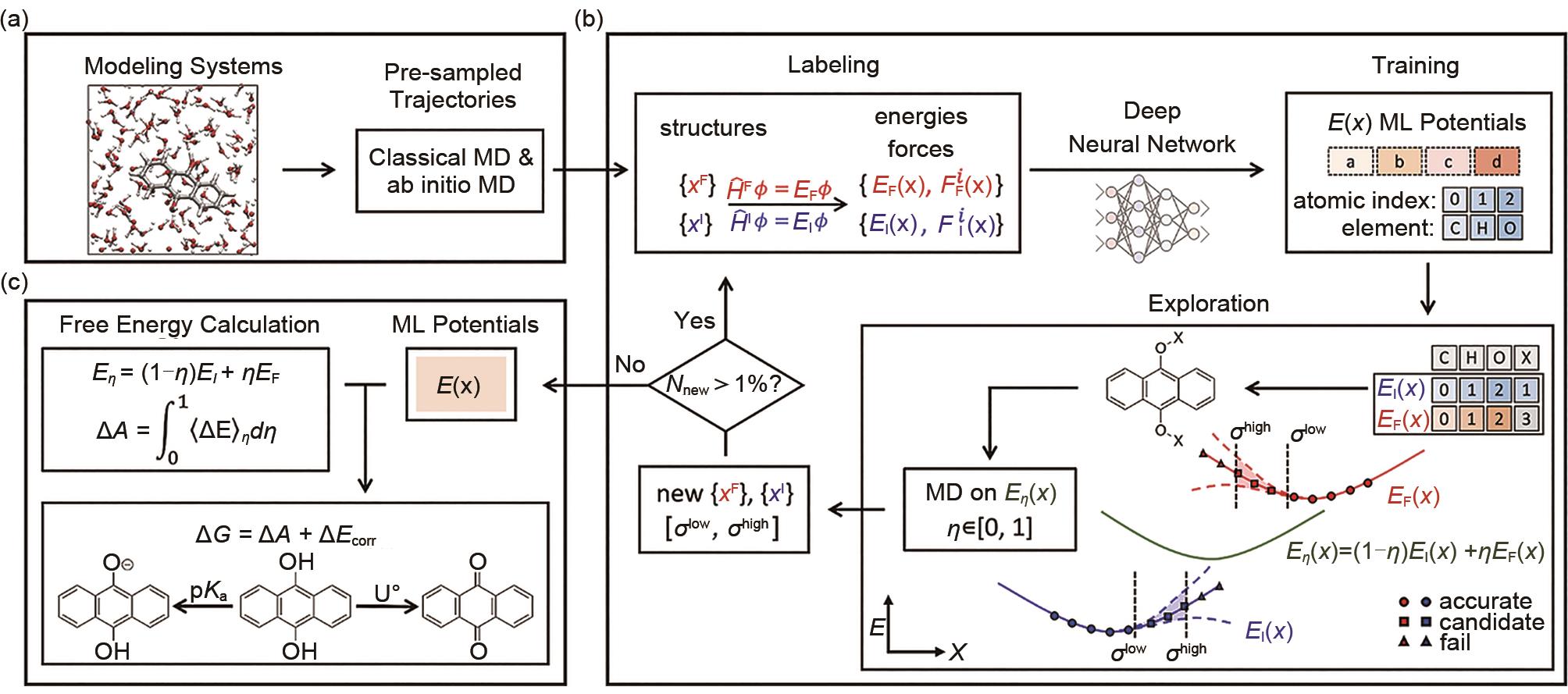
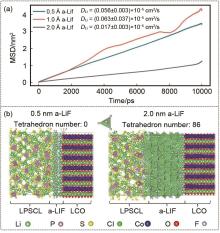
图15
(a) 不同厚度的α-LiF涂层区域内Li离子的均方位移(MSD)随模拟时间的变化及获得的扩散系数。(b) 0.5 nm和2.0 nm层厚度的LiF的原子堆积形貌。规则的Li-F四面体数和P-S四面体也被显示[72]"

| 1 | ZHANG L F, HAN J Q, WANG H, et al. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics[J]. Physical Review Letters, 2018, 120(14): 143001. DOI: 10.1103/PhysRevLett.120.143001. |
| 2 | BEHLER J, PARRINELLO M. Generalized neural-network representation of high-dimensional potential-energy surfaces[J]. Physical Review Letters, 2007, 98(14): 146401. DOI: 10.1103/PhysRevLett.98.146401. |
| 3 | SCHütt K, KINDERMANS P J, SAUCEDA FELIX H E, et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions[J]. Advances in neural information processing systems, 2017, 30. |
| 4 | ZHANG D, BI H R, DAI F Z, et al. Pretraining of attention-based deep learning potential model for molecular simulation[J]. NPJ Computational Materials, 2024, 10: 94. DOI: 10.1038/s41524-024-01278-7. |
| 5 | ZHANG D, LIU X Z J, ZHANG X Y, et al. DPA-2: Towards a universal large atomic model for molecular and materials simulation[J]. arXiv, 2024. DOI: arxiv-2312.15492. |
| 6 | JIA W L, WANG H, CHEN M H, et al. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning[C]//SC20: International Conference for High Performance Computing, Networking, Storage and Analysis. IEEE, 2020: 1-14. DOI: 10.1109/SC41405.2020.00009. |
| 7 | LI J H, WEI N, LI J L, et al. Physicochemical properties of cathode materials for failed lithium iron phosphate batteries[J]. China Powder Science and Technology, 2022, 28(6). |
| 8 | CHEN K, LIAO Q, LIU K, et al. Capacity degradation prediction of lithium-ion battery based on artificial bee colony and multi-kernel support vector regression[J]. Journal of Energy Storage, 2023, 72: 108160. DOI: 10.1016/j.est.2023.108160. |
| 9 | CHEN K, ZHOU S Y, LIU K, et al. State of charge estimation for lithium-ion battery based on whale optimization algorithm and multi-kernel relevance vector machine[J]. 2023, 158(10): 104110. DOI: 10.1063/5.0139376. |
| 10 | HUANG J X, ZHANG L F, WANG H, et al. Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors[J]. 2021, 154(9): 094703. DOI: 10. 1063/5.0041849. |
| 11 | WANG F, MA Z B, CHENG J. Accelerating computation of acidity constants and redox potentials for aqueous organic redox flow batteries by machine learning potential-based molecular dynamics[J]. Journal of the American Chemical Society, 2024, 146(21): 14566-14575. DOI: 10.1021/jacs.4c01221. |
| 12 | KRESSE G, FURTHMÜLLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1): 15-50. DOI: 10.1016/0927-0256(96)00008-0. |
| 13 | VANDEVONDELE J, KRACK M, MOHAMED F, et al. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach[J]. Computer Physics Communications, 2005, 167(2): 103-128. DOI: 10.1016/j.cpc. 2004.12.014. |
| 14 | FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09, Revision E.01[M]. Gaussian, Inc., Wallingford CT, 2013. |
| 15 | GIANNOZZI P, BARONI S, BONINI N, et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials[J]. Journal of Physics Condensed Matter, 2009, 21(39): 395502. DOI: 10.1088/0953-8984/21/39/395502. |
| 16 | WANG H, ZHANG L F, HAN J Q, et al. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics[J]. Computer Physics Communications, 2018, 228: 178-184. DOI: 10.1016/j.cpc.2018.03.016. |
| 17 | THOMPSON A P, AKTULGA H M, BERGER R, et al. LAMMPS-A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales[J]. Computer Physics Communications, 2022, 271: 108171. DOI: 10.1016/j.cpc. 2021.108171. |
| 18 | LARSEN A H, MORTENSEN J J, BLOMQVIST J, et al. The atomic simulation environment-A Python library for working with atoms[J]. Journal of Physics Condensed Matter, 2017, 29(27): 273002. DOI: 10.1088/1361-648X/aa680e. |
| 19 | CERIOTTI M, MORE J, MANOLOPOULOS D E. I-PI: A Python interface for ab initio path integral molecular dynamics simulations[J]. Computer Physics Communications, 2014, 185(3): 1019-1026. DOI: 10.1016/j.cpc.2013.10.027. |
| 20 | ABRAHAM M J, MURTOLA T, SCHULZ R, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers[J]. SoftwareX, 2015, 1: 19-25. DOI: 10.1016/j.softx.2015.06.001. |
| 21 | HUA H M, WANG F, WANG F, et al. Machine learning molecular dynamics insight into high interface stability and fast kinetics of low-cost magnesium chloride amine electrolyte for rechargeable magnesium batteries[J]. Energy Storage Materials, 2024, 70: 103470. DOI: 10.1016/j.ensm.2024.103470. |
| 22 | WANG J J, SHEN H, YANG R Y, et al. A deep learning interatomic potential developed for atomistic simulation of carbon materials[J]. Carbon, 2022, 186: 1-8. DOI: 10.1016/j.carbon. 2021.09.062. |
| 23 | OBEID M M, LIU J H, DU P H, et al. A 3D metallic porous sulfurized carbon anode identified by global structure search for Na-ion batteries with fast diffusion kinetics[J]. Journal of Energy Storage, 2024, 82: 110587. DOI: 10.1016/j.est.2024.110587. |
| 24 | XU N, SHI Y, HE Y, et al. A deep-learning potential for crystalline and amorphous Li-Si alloys[J]. The Journal of Physical Chemistry C, 2020, 124(30): 16278-16288. DOI: 10.1021/acs.jpcc.0c03333. |
| 25 | FU F J, WANG X X, ZHANG L F, et al. Unraveling the atomic-scale mechanism of phase transformations and structural evolutions during (de)lithiation in Si anodes[J]. Advanced Functional Materials, 2023, 33(37): 2303936. DOI: 10.1002/adfm.202303936. |
| 26 | JIAO J Y, LAI G M, ZHAO L, et al. Self-healing mechanism of lithium in lithium metal[J]. Advanced Science, 2022, 9(12): e2105574. DOI: 10.1002/advs.202105574. |
| 27 | PHUTHI M K, YAO A M, BATZNER S, et al. Accurate surface and finite-temperature bulk properties of lithium metal at large scales using machine learning interaction potentials[J]. ACS Omega, 2024, 9(9): 10904-10912. DOI: 10.1021/acsomega.3c10014. |
| 28 | LAI G M, JIAO J Y, FANG C, et al. A deep neural network interface potential for Li-Cu systems[J]. Advanced Materials Interfaces, 2022, 9(27): 2201346. DOI: 10.1002/admi.202201346. |
| 29 | LAI G M, JIAO J Y, FANG C, et al. The mechanism of Li deposition on the Cu substrates in the anode-free Li metal batteries[J]. Small, 2023, 19(3): 2205416. DOI: 10.1002/smll. 202205416. |
| 30 | GARCIA J C, GABRIEL J, PAULSON N H, et al. Insights from computational studies on the anisotropic volume change of LixNiO2 at high states of charge (x<0.25)[J]. The Journal of Physical Chemistry C, 2021, 125(49): 27130-27139. DOI: 10. 1021/acs.jpcc.1c08022. |
| 31 | HU T P, DAI F Z, ZHOU G B, et al. Unraveling the dynamic correlations between transition metal migration and the oxygen dimer formation in the highly delithiated LixCoO2 cathode[J]. The Journal of Physical Chemistry Letters, 2023, 14(15): 3677-3684. DOI: 10.1021/acs.jpclett.3c00506. |
| 32 | HU Y C, WANG X X, LI P, et al. Understanding the sluggish and highly variable transport kinetics of lithium ions in LiFePO4[J]. Science China Chemistry, 2023, 66(11): 3297-3306. DOI: 10. 1007/s11426-023-1662-9. |
| 33 | LIN M, LIU X S, XIANG Y X, et al. Unravelling the fast alkali-ion dynamics in paramagnetic battery materials combined with NMR and deep-potential molecular dynamics simulation[J]. Angewandte Chemie International Edtion, 2021, 60(22): 12547-12553. DOI: 10.1002/anie.202102740. |
| 34 | LIN M, XIONG J F, SU M T, et al. A machine learning protocol for revealing ion transport mechanisms from dynamic NMR shifts in paramagnetic battery materials[J]. Chemical Science, 2022, 13(26): 7863-7872. DOI: 10.1039/d2sc01306a. |
| 35 | LIN M, FU R Q, XIANG Y X, et al. Combining NMR and molecular dynamics simulations for revealing the alkali-ion transport in solid-state battery materials[J]. Current Opinion in Electrochemistry, 2022, 35: 101048. DOI: 10.1016/j.coelec.2022.101048. |
| 36 | MARCOLONGO A, BINNINGER T, ZIPOLI F, et al. Simulating diffusion properties of solid-state electrolytes via a neural network potential: Performance and training scheme[J]. ChemSystemsChem, 2020, 2(3). DOI: 10.1002/syst.201900031. |
| 37 | FU Z H, CHEN X, YAO N, et al. The chemical origin of temperature-dependent lithium-ion concerted diffusion in sulfide solid electrolyte Li10GeP2S12[J]. Journal of Energy Chemistry, 2022, 70: 59-66. DOI: 10.1016/j.jechem.2022.01.018. |
| 38 | MIYAGAWA T, KRISHNAN N, GRUMET M, et al. Accurate description of ion migration in solid-state ion conductors from machine-learning molecular dynamics[J]. Journal of Materials Chemistry A, 2024, 12(19): 11344-11361. DOI: 10.1039/D4TA00452C. |
| 39 | GIGLI L, TISI D, GRASSELLI F, et al. Mechanism of charge transport in lithium thiophosphate[J]. Chemistry of Materials, 2024, 36(3): 1482-1496. DOI: 10.1021/acs.chemmater.3c02726. |
| 40 | GUPTA M K, DING J X, OSTI N C, et al. Fast Na diffusion and anharmonic phonon dynamics in superionic Na3PS4[J]. Energy & Environmental Science, 2021, 14(12): 6554-6563. DOI: 10.1039/D1EE01509E. |
| 41 | ZHANG R Y, XU S F, WANG L Y, et al. Theoretical study on ion diffusion mechanism in W-doped K3SbS4 as solid-state electrolyte for K-ion batteries[J]. Inorganic Chemistry, 2024, 63(15): 6743-6751. DOI: 10.1021/acs.inorgchem.4c00074. |
| 42 | LEE J W, KIM J H, KIM J S, et al. Design of multicomponent argyrodite based on a mixed oxidation state as promising solid-state electrolyte using moment tensor potentials[J]. Journal of Materials Chemistry A, 2024, 12(12): 7272-7278. DOI: 10.1039/D4TA00361F. |
| 43 | GUPTA M K, KUMAR S, MITTAL R, et al. Soft-phonon anharmonicity, floppy modes, and Na diffusion in Na3FY(Y=S, Se, Te): Ab initio and machine-learned molecular dynamics simulations[J]. Physical Review B, 2022, 106: 014311. DOI: 10. 1103/physrevb.106.014311. |
| 44 | SELVARAJ S C, KOVERGA V, NGO A T. Exploring Li-ion transport properties of Li3TiCl6: A machine learning molecular dynamics study[J]. Journal of the Electrochemical Society, 2024, 171(5): 050544. DOI: 10.1149/1945-7111/ad4ac9. |
| 45 | ZHANG Z, MA Z Y, PEI Y. Li ion diffusion behavior of Li3OCl solid-state electrolytes with different defect structures: Insights from the deep potential model[J]. Physical Chemistry Chemical Physics, 2023, 25(19): 13297-13307. DOI: 10.1039/d2cp06073f. |
| 46 | LI H X, ZHOU X Y, WANG Y C, et al. Theoretical study of Na+ transport in the solid-state electrolyte Na3OBr based on deep potential molecular dynamics[J]. Inorganic Chemistry Frontiers, 2021, 8(2): 425-432. DOI: 10.1039/D0QI00921K. |
| 47 | LIU J H, WANG S, KAWAZOE Y, et al. A new spinel chloride solid electrolyte with high ionic conductivity and stability for Na-ion batteries[J]. ACS Materials Letters, 2023, 5(4): 1009-1017. DOI: 10.1021/acsmaterialslett.3c00119. |
| 48 | YOU Y W, ZHANG D X, WU F L, et al. Principal component analysis enables the design of deep learning potential precisely capturing LLZO phase transitions[J]. NPJ Computational Materials, 2024, 10: 57. DOI: 10.1038/s41524-024-01240-7. |
| 49 | BALYAKIN I A, VLASOV M I, PERSHINA S V, et al. Neural network molecular dynamics study of LiGe2(PO4)3: Investigation of structure[J]. Computational Materials Science, 2024, 239: 112979. DOI: 10.1016/j.commatsci.2024.112979. |
| 50 | ZHOU R, LUO K, MARTIN S W, et al. Insights into lithium sulfide glass electrolyte structures and ionic conductivity via machine learning force field simulations[J]. ACS Applied Materials & Interfaces, 2024, 16(15): 18874-18887. DOI: 10.1021/acsami. 4c00618. |
| 51 | GUPTA S, YANG X C, CEDER G. What dictates soft clay-like lithium superionic conductor formation from rigid salts mixture[J]. Nature Communications, 2023, 14(1): 6884. DOI: 10.1038/s41467-023-42538-2. |
| 52 | YANG X C, GUPTA S, CHEN Y, et al. Fast room-temperature Mg-ion conduction in clay-like halide glassy electrolytes[J]. Advanced Energy Materials, 2024, 14(26): 2400163. DOI: 10.1002/aenm. 202400163. |
| 53 | ZHANG D X, YOU Y W, WU F L, et al. Exploring the relationship between composition and Li-ion conductivity in the amorphous Li-La-Zr-O system[J]. ACS Materials Letters, 2024, 6(5): 1849-1855. DOI: 10.1021/acsmaterialslett.3c01558. |
| 54 | DAI T, WU S Y, LU Y X, et al. Inorganic glass electrolytes with polymer-like viscoelasticity[J]. Nature Energy, 2023, 8: 1221-1228. DOI: 10.1038/s41560-023-01356-y. |
| 55 | WANG F, CHENG J. Automated workflow for computation of redox potentials, acidity constants, and solvation free energies accelerated by machine learning[J]. Journal of Chemical Physics, 2022, 157(2): 024103. DOI: 10.1063/5.0098330. |
| 56 | WANG F, SUN Y, CHENG J. Switching of redox levels leads to high reductive stability in water-in-salt electrolytes[J]. Journal of the American Chemical Society, 2023, 145(7): 4056-4064. DOI: 10.1021/jacs.2c11793. |
| 57 | ZHANG C Y, YUE S W, PANAGIOTOPOULOS A Z, et al. Why dissolving salt in water decreases its dielectric permittivity[J]. Physical Review Letters, 2023, 131(7): 076801. DOI: 10.1103/PhysRevLett.131.076801. |
| 58 | PANAGIOTOPOULOS A Z, YUE S W. Dynamics of aqueous electrolyte solutions: Challenges for simulations[J]. The Journal of Physical Chemistry B, 2023, 127(2): 430-437. DOI: 10.1021/acs.jpcb.2c07477. |
| 59 | ZHU D, SHENG L, HU T P, et al. Investigation of the degradation of LiPF6 - in polar solvents through deep potential molecular dynamics[J]. The Journal of Physical Chemistry Letters, 2024, 15(15): 4024-4030. DOI: 10.1021/acs.jpclett.4c00575. |
| 60 | LING Y L, LI K, WANG M, et al. Revisiting the structure, interaction, and dynamical property of ionic liquid from the deep learning force field[J]. Journal of Power Sources, 2023, 555: 232350. DOI: 10.1016/j.jpowsour.2022.232350. |
| 61 | XU M Y, ZHU T, ZHANG J Z H. Molecular dynamics simulation of zinc ion in water with an ab initio based neural network potential[J]. The Journal of Physical Chemistry A, 2019, 123(30): 6587-6595. DOI: 10.1021/acs.jpca.9b04087. |
| 62 | LIU J C, LIU R X, CAO Y, et al. Solvation structures of calcium and magnesium ions in water with the presence of hydroxide: A study by deep potential molecular dynamics[J]. Physical Chemistry Chemical Physics, 2023, 25(2): 983-993. DOI: 10. 1039/d2cp04105g. |
| 63 | WANG F, CHENG J. Understanding the solvation structures of glyme-based electrolytes by machine learning molecular dynamics[J]. Chinese Journal of Structural Chemistry, 2023, 42(9): 100061. DOI: 10.1016/j.cjsc.2023.100061. |
| 64 | XU T R, LI X J, WANG Y, et al. Development of deep potentials of molten MgCl2-NaCl and MgCl2-KCl salts driven by machine learning[J]. ACS Applied Materials & Interfaces, 2023. DOI: 10.1021/acsami.2c19272. |
| 65 | QI S M, BO T, ZHANG L, et al. Machine-learning-driven simulations on microstructure, thermodynamic properties, and transport properties of LiCl-KCl-LiF molten salt[J]. Artificial Intelligence Chemistry, 2024, 2(1): 100027. DOI: 10.1016/j.aichem.2023.100027. |
| 66 | LE J B, CHEN A, LI L, et al. Modeling electrified Pt(111)-Had/water interfaces from ab initio molecular dynamics[J]. JACS Au, 2021, 1(5): 569-577. DOI: 10.1021/jacsau.1c00108. |
| 67 | LE J B, FAN Q Y, LI J Q, et al. Molecular origin of negative component of Helmholtz capacitance at electrified Pt(111)/water interface[J]. Science Advances, 2020, 6(41): eabb1219. DOI: 10.1126/sciadv.abb1219. |
| 68 | LI J Q, SUN Y, CHENG J. Theoretical investigation on water adsorption conformations at aqueous anatase TiO2/water interfaces[J]. Journal of Materials Chemistry A, 2023, 11(2): 943-952. DOI: 10.1039/D2TA07994A. |
| 69 | ZHUANG Y B, CHENG J. Deciphering the anomalous acidic tendency of terminal water at rutile(110)-water interfaces[J]. The Journal of Physical Chemistry C, 2023, 127(22): 10532-10540. DOI: 10.1021/acs.jpcc.3c01870. |
| 70 | BIN JASSAR M, MICHEL C, ABADA S, et al. A perspective on the molecular modeling of electrolyte decomposition reactions for solid electrolyte interphase growth in lithium-ion batteries[J]. Advanced Functional Materials, 2024, 34(30): 2313188. DOI: 10.1002/adfm.202313188. |
| 71 | REN F C, WU Y Q, ZUO W H, et al. Visualizing the SEI formation between lithium metal and solid-state electrolyte[J]. Energy & Environmental Science, 2024, 17(8): 2743-2752. DOI: 10.1039/D3EE03536K. |
| 72 | HU T P, XU L H, DAI F Z, et al. Impact of amorphous LiF coating layers on cathode-electrolyte interfaces in solid-state batteries[J]. Advanced Functional Materials, 2024: 2402993. DOI: 10.1002/adfm.202402993. |
| 73 | HU T P, TIAN J X, DAI F Z, et al. Impact of the local environment on Li ion transport in inorganic components of solid electrolyte interphases[J]. Journal of the American Chemical Society, 2023, 145(2): 1327-1333. DOI: 10.1021/jacs.2c11521. |
| 74 | CHEN M, GUO G C, HE L. Systematically improvable optimized atomic basis sets for ab initio calculations [J]. Journal of Physics: Condensed Matter, 2010, 22(44): 445501. |
| [1] | 陆继忠, 彭思敏, 李晓宇. 基于多特征量分析和LSTM-XGBoost模型的锂离子电池SOH估计方法[J]. 储能科学与技术, 2024, 13(9): 2972-2982. |
| [2] | 管鸿盛, 钱诚, 孙博, 任羿. 贫数据条件下锂离子电池容量退化轨迹预测方法[J]. 储能科学与技术, 2024, 13(9): 3084-3093. |
| [3] | 柯学, 洪华伟, 郑鹏, 李智诚, 范培潇, 杨军, 郭宇铮, 蒯春光. 基于多时间尺度建模自动特征提取和通道注意力机制的锂离子电池健康状态估计[J]. 储能科学与技术, 2024, 13(9): 3059-3071. |
| [4] | 许晶, 王宇琦, 符晓, 杨其凡, 连景臣, 王力奇, 肖睿娟. 基于大数据的电池新材料设计[J]. 储能科学与技术, 2024, 13(9): 2920-2932. |
| [5] | 李从心, 岳美玲, 李昕彤, 熊庆辉, 刘孝艳. 基于条件神经网络的质子交换膜燃料电池的老化性能预测[J]. 储能科学与技术, 2024, 13(9): 3094-3102. |
| [6] | 何婷, 乔俊强, 吴国栋. 基于GRU算法的弃电量预测及电-氢混合储能系统的运行优化[J]. 储能科学与技术, 2024, 13(5): 1731-1740. |
| [7] | 杨立杰. 相变储能材料在建筑工程建设中的应用[J]. 储能科学与技术, 2024, 13(5): 1471-1473. |
| [8] | 曾其权, 罗马吉, 杨印龙, 黄庆泽. 基于LSTM-UPF混合驱动方法的燃料电池寿命预测[J]. 储能科学与技术, 2024, 13(3): 963-970. |
| [9] | 张雪丽, 孙伟清, 郑君华. 聚氨酯型固-固相变储能材料对沥青调温效果的影响研究[J]. 储能科学与技术, 2024, 13(3): 841-843. |
| [10] | 裴雯. 海上物流中相变储能材料的制备与热物性能分析[J]. 储能科学与技术, 2024, 13(3): 844-846. |
| [11] | 牛红培. 相变储能材料在节能建筑设计中的应用[J]. 储能科学与技术, 2024, 13(3): 847-849. |
| [12] | 陈欣, 李云伍, 梁新成, 李法霖, 张志冬. 基于模态分解的Transformer-GRU联合电池健康状态估计[J]. 储能科学与技术, 2023, 12(9): 2927-2936. |
| [13] | 陈智伟, 张维戈, 张珺玮, 张言茹. 基于视觉特征的动力电池组综合健康评估及分筛方法[J]. 储能科学与技术, 2023, 12(7): 2211-2219. |
| [14] | 张宇波, 王有元, 黄洞宁, 王子懿, 陈伟根. 面向变工况条件的锂离子电池寿命退化预测方法[J]. 储能科学与技术, 2023, 12(7): 2238-2245. |
| [15] | 管鸿盛, 钱诚, 徐炳辉, 孙博, 任羿. 融合自注意力机制与门控循环单元网络的宽工况锂离子电池SOC估计[J]. 储能科学与技术, 2023, 12(7): 2229-2237. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
