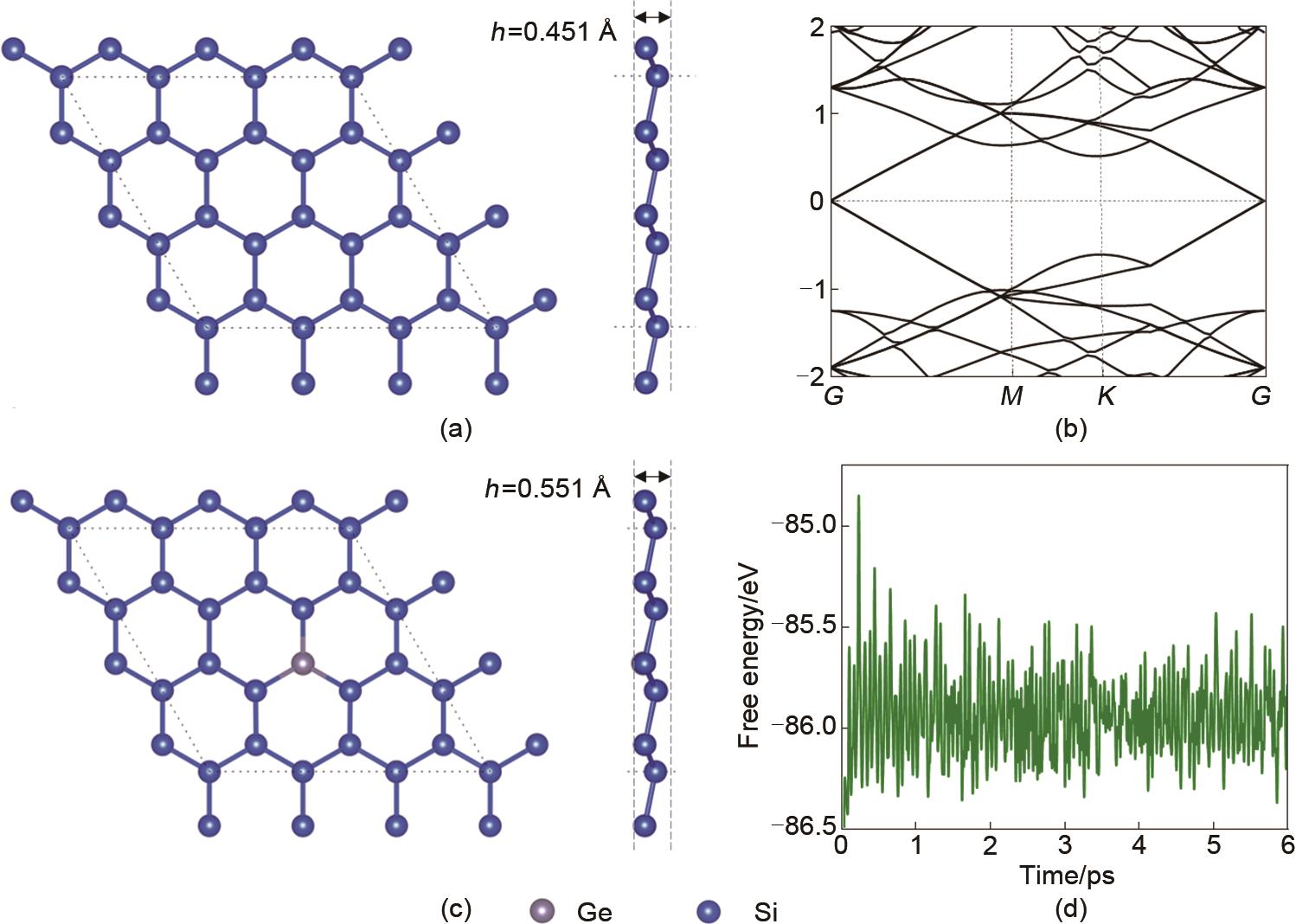
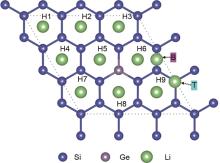
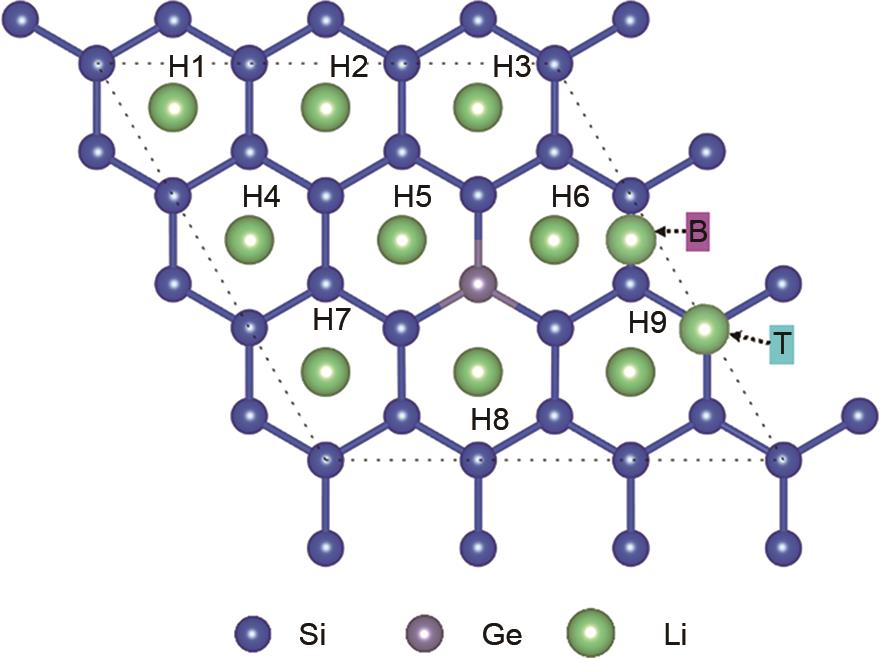
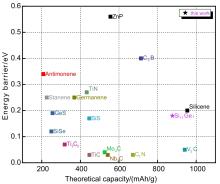
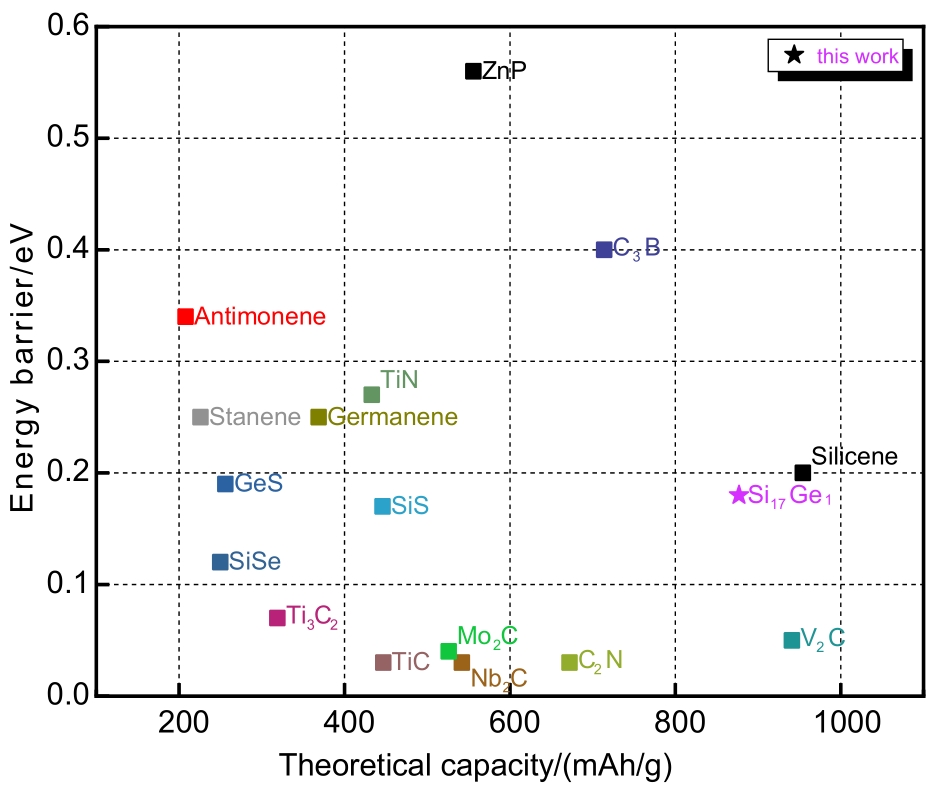
| 1 |
NZEREOGU P U, OMAH A D, EZEMA F I, et al. Anode materials for lithium-ion batteries: A review[J]. Applied Surface Science Advances, 2022, 9: 100233.
|
| 2 |
LIU K, YANG S L, LUO L Q, et al. From spent graphite to recycle graphite anode for high-performance lithium ion batteries and sodium ion batteries[J]. Electrochimica Acta, 2020, 356: 136856.
|
| 3 |
ROJAEE R, SHAHBAZIAN-YASSAR R. Two-dimensional materials to address the lithium battery challenges[J]. ACS Nano, 2020, 14(3): 2628-2658.
|
| 4 |
ZHUANG J C, XU X, PELECKIS G, et al. Silicene: A promising anode for lithium-ion batteries[J]. Advanced Materials, 2017, 29(48): 1606716.
|
| 5 |
TRITSARIS G A, KAXIRAS E, MENG S, et al. Adsorption and diffusion of lithium on layered silicon for Li-ion storage[J]. Nano Letters, 2013, 13(5): 2258-2263.
|
| 6 |
WAN W H, ZHANG Q F, CUI Y, et al. First principles study of lithium insertion in bulk silicon[J]. Journal of Physics Condensed Matter: an Institute of Physics Journal, 2010, 22(41): 415501.
|
| 7 |
SHI L, ZHAO T S, XU A, et al. Ab initio prediction of a silicene and graphene heterostructure as an anode material for Li- and Na-ion batteries[J]. Journal of Materials Chemistry A, 2016, 4(42): 16377-16382.
|
| 8 |
LV X S, WEI W, HUANG B B, et al. Achieving high energy density for lithium-ion battery anodes by Si/C nanostructure design[J]. Journal of Materials Chemistry A, 2019, 7(5): 2165-2171.
|
| 9 |
SHUKLA V, ARAUJO R B, JENA N K, et al. The curious case of two dimensional Si2BN: A high-capacity battery anode material[J]. Nano Energy, 2017, 41: 251-260.
|
| 10 |
LI H, HOU J H, JIANG D Y. 2D Si3N as a promising anode material for Li/Na-ion batteries from first-principles study[J]. Journal of Electronic Materials, 2020, 49(7): 4180-4185.
|
| 11 |
ZIA A, CAI Z P, NAVEED A B, et al. MXene, silicene and germanene: Preparation and energy storage applications[J]. Materials Today Energy, 2022, 30: 101144.
|
| 12 |
SANNYAL A, AHN Y, JANG J. First-principles study on the two-dimensional siligene (2D SiGe) as an anode material of an alkali metal ion battery[J]. Computational Materials Science, 2019, 165: 121-128.
|
| 13 |
SONG J, JIANG M J, WAN C, et al. Defective graphene/SiGe heterostructures as anodes of Li-ion batteries: A first-principles calculation study[J]. Physical Chemistry Chemical Physics: PCCP, 2022, 25(1): 617-624.
|
| 14 |
CHEN X, LOAIZA L C, MONCONDUIT L, et al. 2D silicon-germanium-layered materials as anodes for Li-ion batteries[J]. ACS Applied Energy Materials, 2021, 4(11): 12552-12561.
|
| 15 |
EHRLICH S, MOELLMANN J, RECKIEN W, et al. System-dependent dispersion coefficients for the DFT-D3 treatment of adsorption processes on ionic surfaces[J]. Chemphyschem: a European Journal of Chemical Physics and Physical Chemistry, 2011, 12(17): 3414-3420.
|
| 16 |
KUTNER R. Chemical diffusion in the lattice gas of non-interacting particles[J]. Physics Letters A, 1981, 81(4): 239-240.
|
| 17 |
HUANG J, CHEN H J, WU M S, et al. First-principles calculation of lithium adsorption and diffusion on silicene[J]. Chinese Physics Letters, 2013, 30(1): 017103.
|
| 18 |
HU J P, OUYANG C Y, YANG S A, et al. Germagraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations[J]. Nanoscale Horizons, 2019, 4(2): 457-463.
|
| 19 |
WANG H, WU M, LEI X, et al. Siligraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations [J]. Nano Energy, 2018, 49: 67-76.
|
| 20 |
GAO X, LU W Q, XU J. Insights into the Li diffusion mechanism in Si/C composite anodes for lithium-ion batteries[J]. ACS Applied Materials & Interfaces, 2021, 13(18): 21362-21370.
|
| 21 |
FAN X F, ZHENG W T, KUO J L. Adsorption and diffusion of Li on pristine and defective graphene[J]. ACS Applied Materials & Interfaces, 2012, 4(5): 2432-2438.
|
| 22 |
SETIADI J, ARNOLD M D, FORD M J. Li-ion adsorption and diffusion on two-dimensional silicon with defects: A first principles study[J]. ACS Applied Materials & Interfaces, 2013, 5(21): 10690-10695.
|
| 23 |
MOMENI M J, MOUSAVI-KHOSHDEL M, TARGHOLI E. First-principles investigation of adsorption and diffusion of Li on doped silicenes: Prospective materials for lithium-ion batteries[J]. Materials Chemistry and Physics, 2017, 192: 125-130.
|
| 24 |
DAS D, KIM S, LEE K R, et al. Li diffusion through doped and defected graphene[J]. Physical Chemistry Chemical Physics: PCCP, 2013, 15(36): 15128-15134.
|
| 25 |
WANG Z H, RATVIK A P, GRANDE T, et al. Diffusion of alkali metals in the first stage graphite intercalation compounds by vdW-DFT calculations[J]. RSC Advances, 2015, 5(21): 15985-15992.
|
| 26 |
BAHARI Y, MORTAZAVI B, RAJABPOUR A, et al. Application of two-dimensional materials as anodes for rechargeable metal-ion batteries: A comprehensive perspective from density functional theory simulations[J]. Energy Storage Materials, 2021, 35: 203-282.
|
| 27 |
ZHOU Y G. MX (M=Ge, Sn; X=S, Se) sheets: Theoretical prediction of new promising electrode materials for Li ion batteries[J]. Journal of Materials Chemistry A, 2016, 4(28): 10906-10913.
|
| 28 |
HU J P, XU B, OUYANG C, et al. Investigations on V2C and V2CX2 (X=F, OH) monolayer as a promising anode material for Li ion batteries from first-principles calculations[J]. The Journal of Physical Chemistry C, 2014, 118(42): 24274-24281.
|
| 29 |
HU J P, XU B, OUYANG C Y, et al. Investigations on Nb2C monolayer as promising anode material for Li or non-Li ion batteries from first-principles calculations[J]. RSC Advances, 2016, 6(33): 27467-27474.
|
| 30 |
ÇAKıR D, SEVIK C, GÜLSEREN O, et al. Mo2C as a high capacity anode material: A first-principles study[J]. Journal of Materials Chemistry A, 2016, 4(16): 6029-6035.
|
| 31 |
TANG Q, ZHOU Z, SHEN P W. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X=F, OH) monolayer[J]. Journal of the American Chemical Society, 2012, 134(40): 16909-16916.
|
| 32 |
HANKEL M, SEARLES D J. Lithium storage on carbon nitride, graphenylene and inorganic graphenylene[J]. Physical Chemistry Chemical Physics: PCCP, 2016, 18(21): 14205-14215.
|
| 33 |
LIU Y Y, ARTYUKHOV V I, LIU M J, et al. Feasibility of lithium storage on graphene and its derivatives[J]. The Journal of Physical Chemistry Letters, 2013, 4(10): 1737-1742.
|
| 34 |
ZHANG J H, LIU G, HU H C, et al. Graphene-like carbon-nitrogen materials as anode materials for Li-ion and mg-ion batteries[J]. Applied Surface Science, 2019, 487: 1026-32.
|
| 35 |
MORTAZAVI B, DIANAT A, CUNIBERTI G, et al. Application of silicene, germanene and stanene for Na or Li ion storage: A theoretical investigation[J]. Electrochimica Acta, 2016, 213: 865-870.
|
| 36 |
FAN D, LU S H, GUO Y D, et al. Two-dimensional tetragonal titanium carbide: A high-capacity and high-rate battery material[J]. The Journal of Physical Chemistry C, 2018, 122(27): 15118-15124.
|
| 37 |
KARMAKAR S, CHOWDHURY C, DATTA A. Two-dimensional group IV monochalcogenides: Anode materials for Li-ion batteries[J]. The Journal of Physical Chemistry C, 2016, 120(27): 14522-14530.
|
| 38 |
MORTAZAVI B, BAFEKRY A, SHAHROKHI M, et al. ZnN and ZnP as novel graphene-like materials with high Li-ion storage capacities[J]. Materials Today Energy, 2020, 16: 100392.
|
| 39 |
SENGUPTA A, FRAUENHEIM T. Lithium and sodium adsorption properties of monolayer antimonene[J]. Materials Today Energy, 2017, 5: 347-354.
|

 ), 蒋明杰1, 尚文华1, 李会洁1, 周文俊1, 曾小蔚2
), 蒋明杰1, 尚文华1, 李会洁1, 周文俊1, 曾小蔚2