Energy Storage Science and Technology ›› 2024, Vol. 13 ›› Issue (4): 1293-1301.doi: 10.19799/j.cnki.2095-4239.2023.0669
• Energy Storage Materials and Devices • Previous Articles Next Articles
Jun SONG1( ), Mingjie JIANG1, Wenhua SHANG1, Huijie LI1, Wenjun ZHOU1, Xiaowei ZENG2
), Mingjie JIANG1, Wenhua SHANG1, Huijie LI1, Wenjun ZHOU1, Xiaowei ZENG2
Received:2023-09-26
Revised:2023-10-16
Online:2024-04-26
Published:2024-04-22
Contact:
Jun SONG
E-mail:songjun@zzuli.edu.cn
CLC Number:
Jun SONG, Mingjie JIANG, Wenhua SHANG, Huijie LI, Wenjun ZHOU, Xiaowei ZENG. First-principles study on the effect of Ge doping on the lithium storage behavior of silicene[J]. Energy Storage Science and Technology, 2024, 13(4): 1293-1301.

Fig. 1
Top (a) and side views of the structure of silicene. (b) Band structure, (c) top (a) and side views of the structure of Si17Ge (d) variation of free energies for Si17Ge"
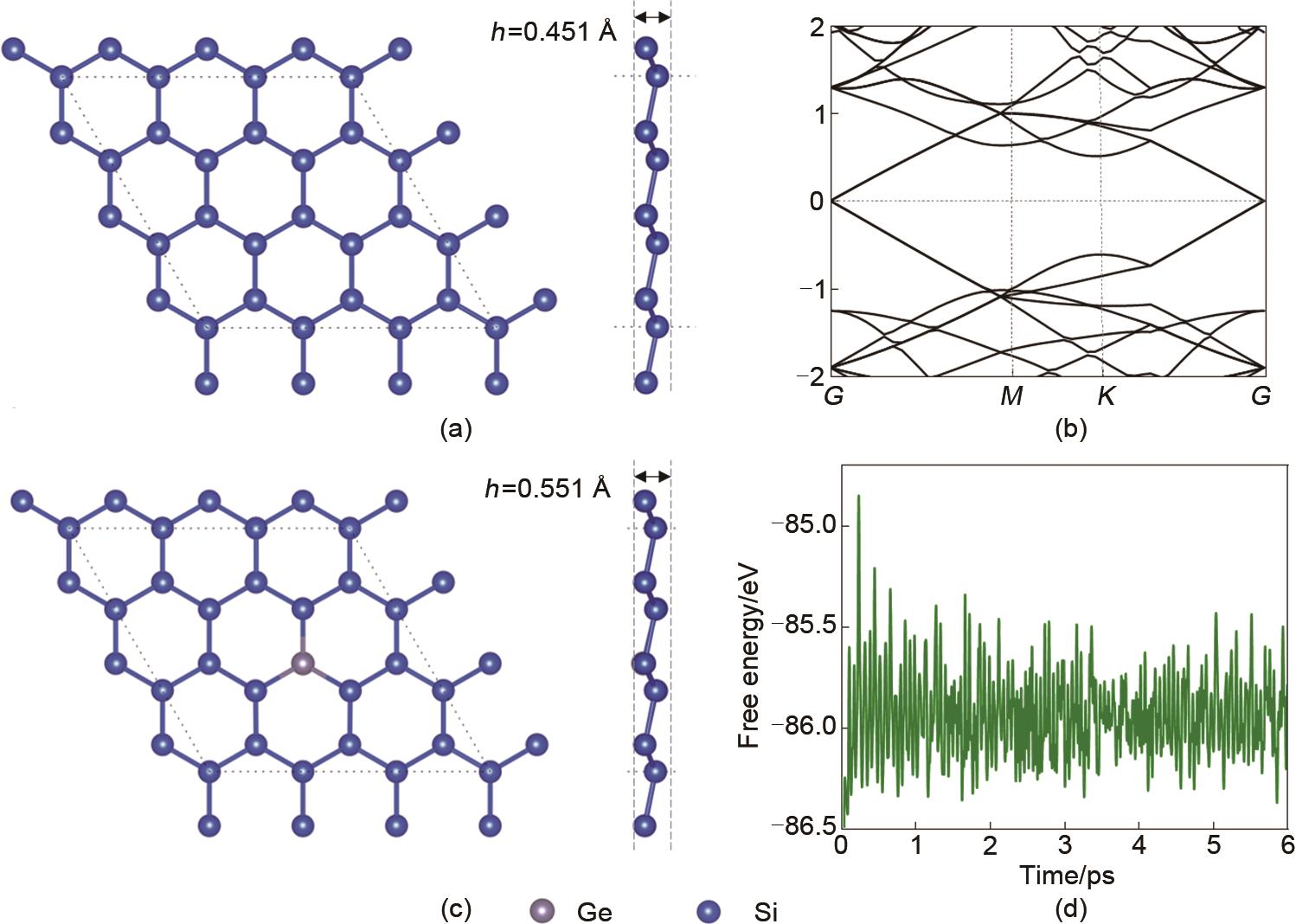
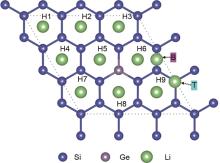
Fig. 2
Top view of possible adsorption sites of lithium ions on Si17Ge"
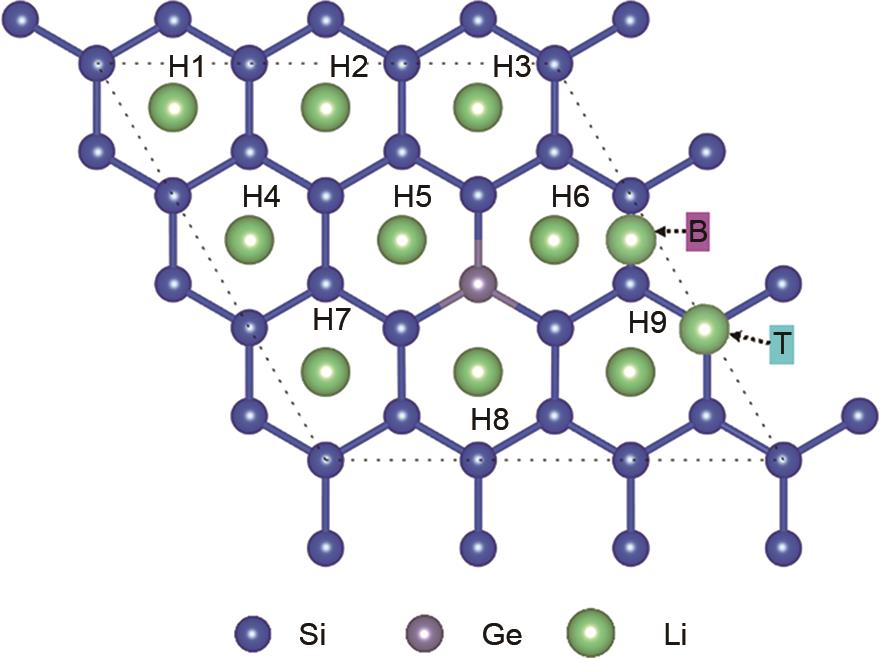
Table 1
The value of adsorption energy and the charge transfer of Li adsorbed on Si17Ge, and the charge transfer (ΔQ) of each element. A positive/negative ΔQ denote a gain/loss of electrons(*:0 means Si17Ge does not absorb lithium atoms)"
| Site | Adsorption energy/eV | Charge state | ||
|---|---|---|---|---|
| ΔQSi | ΔQGe | ΔQLi | ||
| 0* | — | -0.13 | +0.13 | — |
| H1 | -2.09 | +0.32 | +0.20 | -0.52 |
| H2 | -2.09 | +0.38 | +0.19 | -0.57 |
| H3 | -2.09 | +0.00 | +0.54 | -0.54 |
| H4 | -2.09 | +0.46 | +0.07 | -0.53 |
| H5 | -2.10 | +0.34 | +0.15 | -0.49 |
| H6 | -2.10 | +0.26 | +0.27 | -0.53 |
| H7 | -2.09 | +0.26 | +0.24 | -0.50 |
| H8 | -2.10 | +0.20 | +0.24 | -0.43 |
| H9 | -2.09 | -0.11 | +0.61 | -0.50 |

Fig.3
Pathways of Li motion for diffusion calculations across (a) and (b) through the Si17Ge surface, as well as the distribution of energy barriers along the diffusion coordinates"

Fig.4
(a) OCV of different Li concentrations in Si17Ge, (b) the adsorption energy of Li adsorption in Si17Ge"

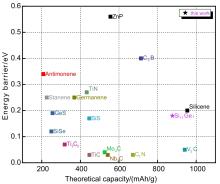
Fig. 5
Maximum storage capacities and diffusion energy barriers of Si17Ge, and other typical two-dimensional anode materials(GeS[27], V2C[28], Nb2C[29], Mo2C[30], Ti3C2[31], g-CN[32], C3B[33], C2N[34], Stanene[35], TiC,TiN[36], SiS, SiSe[37], ZnP[38], Antimonene[39])"
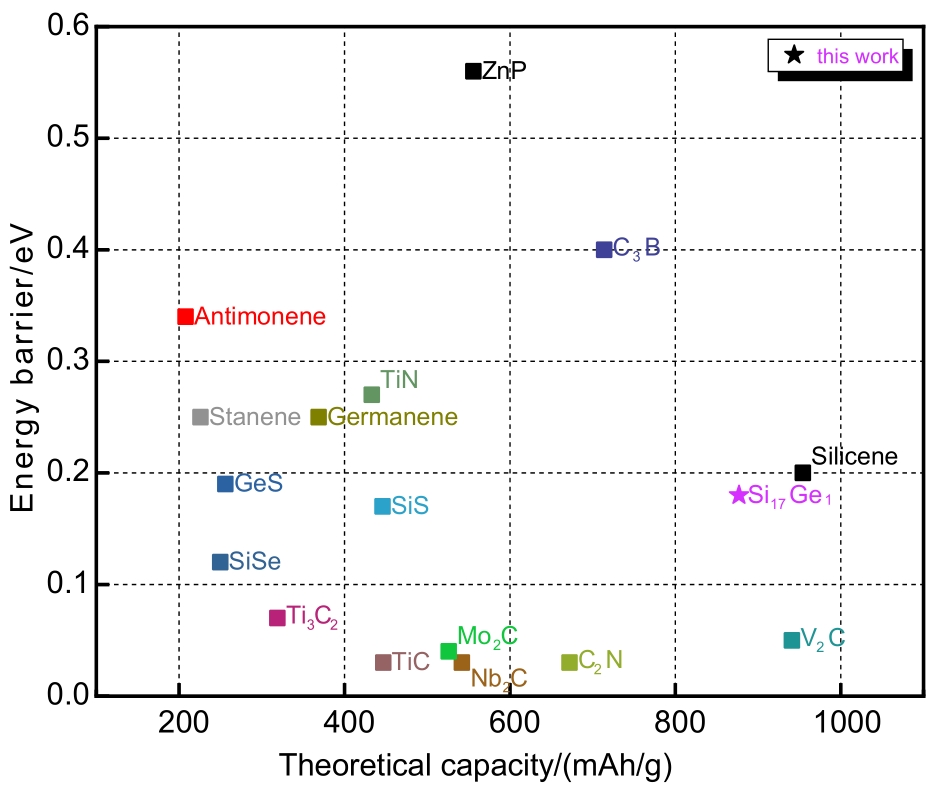

Fig. 6
Density of states for (a)pristine Si17Ge, (b) LiSi17Ge, (c) Li18Si17Ge"
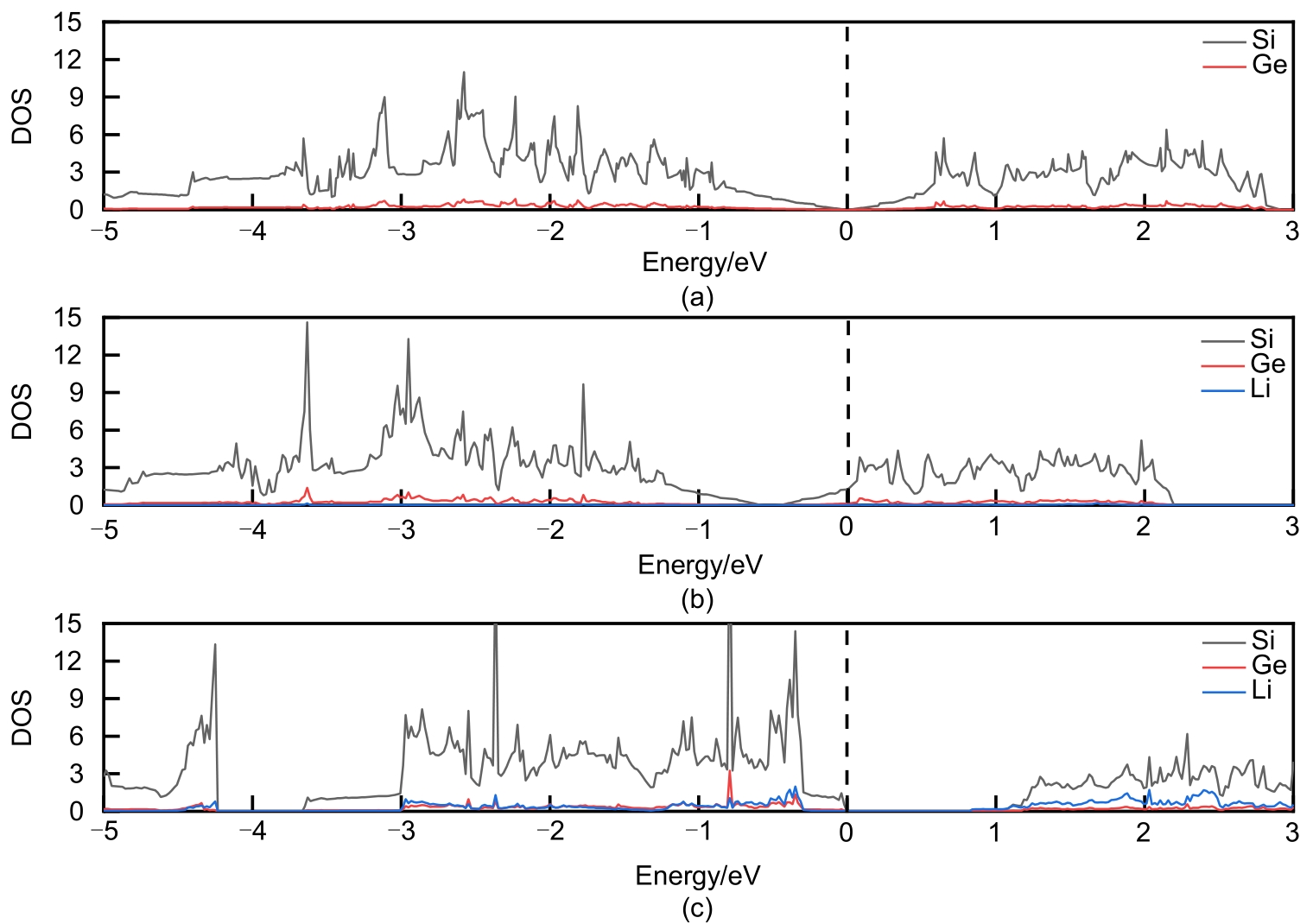
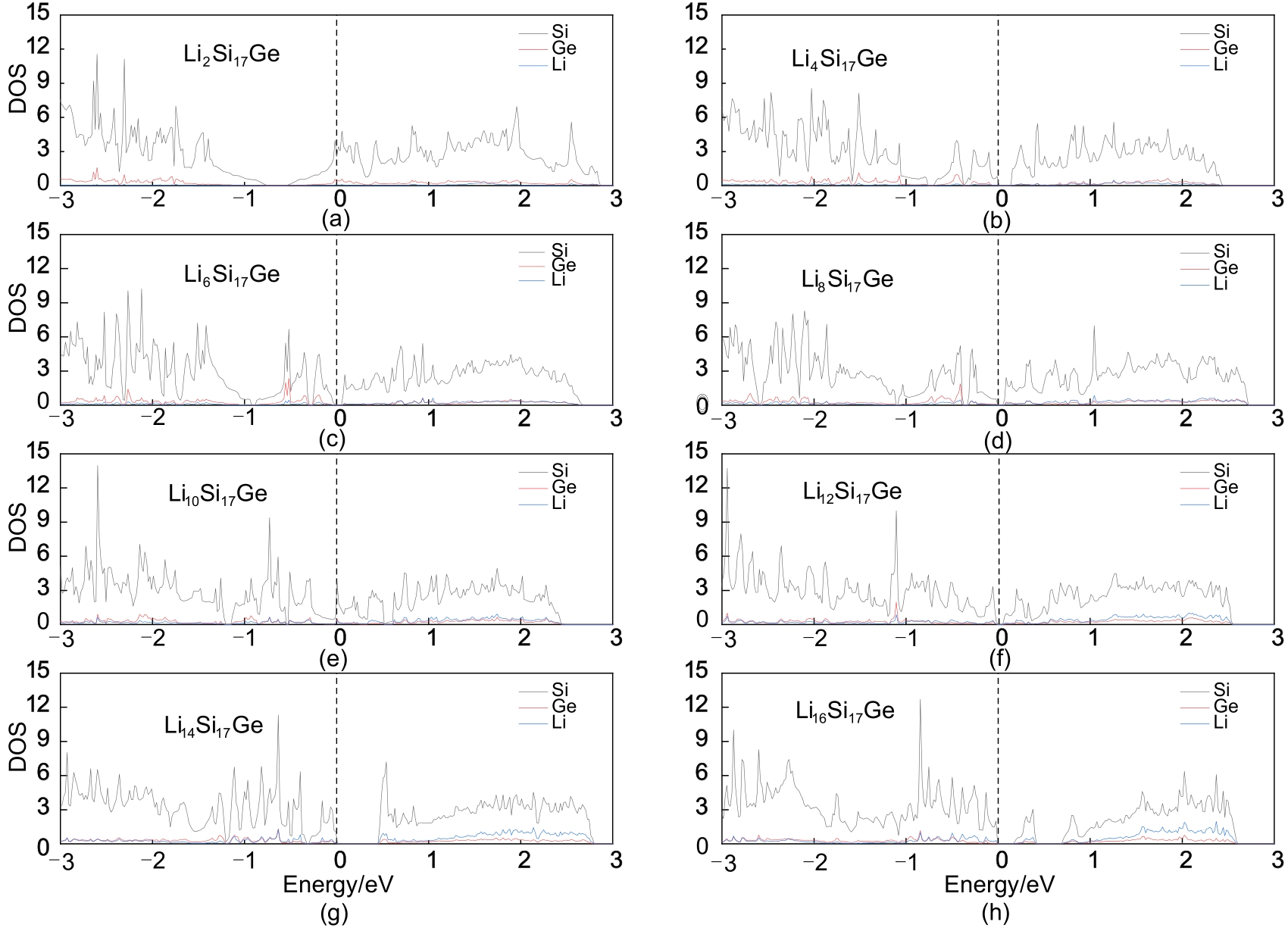
Fig. 7
Density of state when Si17Ge adsorbs (a)2, (b)4, (c)6, (d)8, (e)10, (f)12, (g)14, (h)16 Li atoms"
| 1 | NZEREOGU P U, OMAH A D, EZEMA F I, et al. Anode materials for lithium-ion batteries: A review[J]. Applied Surface Science Advances, 2022, 9: 100233. |
| 2 | LIU K, YANG S L, LUO L Q, et al. From spent graphite to recycle graphite anode for high-performance lithium ion batteries and sodium ion batteries[J]. Electrochimica Acta, 2020, 356: 136856. |
| 3 | ROJAEE R, SHAHBAZIAN-YASSAR R. Two-dimensional materials to address the lithium battery challenges[J]. ACS Nano, 2020, 14(3): 2628-2658. |
| 4 | ZHUANG J C, XU X, PELECKIS G, et al. Silicene: A promising anode for lithium-ion batteries[J]. Advanced Materials, 2017, 29(48): 1606716. |
| 5 | TRITSARIS G A, KAXIRAS E, MENG S, et al. Adsorption and diffusion of lithium on layered silicon for Li-ion storage[J]. Nano Letters, 2013, 13(5): 2258-2263. |
| 6 | WAN W H, ZHANG Q F, CUI Y, et al. First principles study of lithium insertion in bulk silicon[J]. Journal of Physics Condensed Matter: an Institute of Physics Journal, 2010, 22(41): 415501. |
| 7 | SHI L, ZHAO T S, XU A, et al. Ab initio prediction of a silicene and graphene heterostructure as an anode material for Li- and Na-ion batteries[J]. Journal of Materials Chemistry A, 2016, 4(42): 16377-16382. |
| 8 | LV X S, WEI W, HUANG B B, et al. Achieving high energy density for lithium-ion battery anodes by Si/C nanostructure design[J]. Journal of Materials Chemistry A, 2019, 7(5): 2165-2171. |
| 9 | SHUKLA V, ARAUJO R B, JENA N K, et al. The curious case of two dimensional Si2BN: A high-capacity battery anode material[J]. Nano Energy, 2017, 41: 251-260. |
| 10 | LI H, HOU J H, JIANG D Y. 2D Si3N as a promising anode material for Li/Na-ion batteries from first-principles study[J]. Journal of Electronic Materials, 2020, 49(7): 4180-4185. |
| 11 | ZIA A, CAI Z P, NAVEED A B, et al. MXene, silicene and germanene: Preparation and energy storage applications[J]. Materials Today Energy, 2022, 30: 101144. |
| 12 | SANNYAL A, AHN Y, JANG J. First-principles study on the two-dimensional siligene (2D SiGe) as an anode material of an alkali metal ion battery[J]. Computational Materials Science, 2019, 165: 121-128. |
| 13 | SONG J, JIANG M J, WAN C, et al. Defective graphene/SiGe heterostructures as anodes of Li-ion batteries: A first-principles calculation study[J]. Physical Chemistry Chemical Physics: PCCP, 2022, 25(1): 617-624. |
| 14 | CHEN X, LOAIZA L C, MONCONDUIT L, et al. 2D silicon-germanium-layered materials as anodes for Li-ion batteries[J]. ACS Applied Energy Materials, 2021, 4(11): 12552-12561. |
| 15 | EHRLICH S, MOELLMANN J, RECKIEN W, et al. System-dependent dispersion coefficients for the DFT-D3 treatment of adsorption processes on ionic surfaces[J]. Chemphyschem: a European Journal of Chemical Physics and Physical Chemistry, 2011, 12(17): 3414-3420. |
| 16 | KUTNER R. Chemical diffusion in the lattice gas of non-interacting particles[J]. Physics Letters A, 1981, 81(4): 239-240. |
| 17 | HUANG J, CHEN H J, WU M S, et al. First-principles calculation of lithium adsorption and diffusion on silicene[J]. Chinese Physics Letters, 2013, 30(1): 017103. |
| 18 | HU J P, OUYANG C Y, YANG S A, et al. Germagraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations[J]. Nanoscale Horizons, 2019, 4(2): 457-463. |
| 19 | WANG H, WU M, LEI X, et al. Siligraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations [J]. Nano Energy, 2018, 49: 67-76. |
| 20 | GAO X, LU W Q, XU J. Insights into the Li diffusion mechanism in Si/C composite anodes for lithium-ion batteries[J]. ACS Applied Materials & Interfaces, 2021, 13(18): 21362-21370. |
| 21 | FAN X F, ZHENG W T, KUO J L. Adsorption and diffusion of Li on pristine and defective graphene[J]. ACS Applied Materials & Interfaces, 2012, 4(5): 2432-2438. |
| 22 | SETIADI J, ARNOLD M D, FORD M J. Li-ion adsorption and diffusion on two-dimensional silicon with defects: A first principles study[J]. ACS Applied Materials & Interfaces, 2013, 5(21): 10690-10695. |
| 23 | MOMENI M J, MOUSAVI-KHOSHDEL M, TARGHOLI E. First-principles investigation of adsorption and diffusion of Li on doped silicenes: Prospective materials for lithium-ion batteries[J]. Materials Chemistry and Physics, 2017, 192: 125-130. |
| 24 | DAS D, KIM S, LEE K R, et al. Li diffusion through doped and defected graphene[J]. Physical Chemistry Chemical Physics: PCCP, 2013, 15(36): 15128-15134. |
| 25 | WANG Z H, RATVIK A P, GRANDE T, et al. Diffusion of alkali metals in the first stage graphite intercalation compounds by vdW-DFT calculations[J]. RSC Advances, 2015, 5(21): 15985-15992. |
| 26 | BAHARI Y, MORTAZAVI B, RAJABPOUR A, et al. Application of two-dimensional materials as anodes for rechargeable metal-ion batteries: A comprehensive perspective from density functional theory simulations[J]. Energy Storage Materials, 2021, 35: 203-282. |
| 27 | ZHOU Y G. MX (M=Ge, Sn; X=S, Se) sheets: Theoretical prediction of new promising electrode materials for Li ion batteries[J]. Journal of Materials Chemistry A, 2016, 4(28): 10906-10913. |
| 28 | HU J P, XU B, OUYANG C, et al. Investigations on V2C and V2CX2 (X=F, OH) monolayer as a promising anode material for Li ion batteries from first-principles calculations[J]. The Journal of Physical Chemistry C, 2014, 118(42): 24274-24281. |
| 29 | HU J P, XU B, OUYANG C Y, et al. Investigations on Nb2C monolayer as promising anode material for Li or non-Li ion batteries from first-principles calculations[J]. RSC Advances, 2016, 6(33): 27467-27474. |
| 30 | ÇAKıR D, SEVIK C, GÜLSEREN O, et al. Mo2C as a high capacity anode material: A first-principles study[J]. Journal of Materials Chemistry A, 2016, 4(16): 6029-6035. |
| 31 | TANG Q, ZHOU Z, SHEN P W. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X=F, OH) monolayer[J]. Journal of the American Chemical Society, 2012, 134(40): 16909-16916. |
| 32 | HANKEL M, SEARLES D J. Lithium storage on carbon nitride, graphenylene and inorganic graphenylene[J]. Physical Chemistry Chemical Physics: PCCP, 2016, 18(21): 14205-14215. |
| 33 | LIU Y Y, ARTYUKHOV V I, LIU M J, et al. Feasibility of lithium storage on graphene and its derivatives[J]. The Journal of Physical Chemistry Letters, 2013, 4(10): 1737-1742. |
| 34 | ZHANG J H, LIU G, HU H C, et al. Graphene-like carbon-nitrogen materials as anode materials for Li-ion and mg-ion batteries[J]. Applied Surface Science, 2019, 487: 1026-32. |
| 35 | MORTAZAVI B, DIANAT A, CUNIBERTI G, et al. Application of silicene, germanene and stanene for Na or Li ion storage: A theoretical investigation[J]. Electrochimica Acta, 2016, 213: 865-870. |
| 36 | FAN D, LU S H, GUO Y D, et al. Two-dimensional tetragonal titanium carbide: A high-capacity and high-rate battery material[J]. The Journal of Physical Chemistry C, 2018, 122(27): 15118-15124. |
| 37 | KARMAKAR S, CHOWDHURY C, DATTA A. Two-dimensional group IV monochalcogenides: Anode materials for Li-ion batteries[J]. The Journal of Physical Chemistry C, 2016, 120(27): 14522-14530. |
| 38 | MORTAZAVI B, BAFEKRY A, SHAHROKHI M, et al. ZnN and ZnP as novel graphene-like materials with high Li-ion storage capacities[J]. Materials Today Energy, 2020, 16: 100392. |
| 39 | SENGUPTA A, FRAUENHEIM T. Lithium and sodium adsorption properties of monolayer antimonene[J]. Materials Today Energy, 2017, 5: 347-354. |
| [1] | Dalin HU, Panli REN, Changming ZHANG, Mingyang YANG, Zhouguang LU. Improving the cycling performance of LiCoO2 at 4.53 V via in situ co-doping of Al-Y-Zr [J]. Energy Storage Science and Technology, 2024, 13(3): 742-748. |
| [2] | Zhipeng WEN, Kai PAN, Yi WEI, Jiawen GUO, Shanli QIN, Wen JIANG, Lian WU, Huan LIAO. Research progress in lithium manganese iron phosphate cathode material modification [J]. Energy Storage Science and Technology, 2024, 13(3): 770-787. |
| [3] | Jinyu GE, Xianghui MENG, Yongjun QI, Hao SUN, Jianjun LI, Bing ZHOU, Tingting GUI, Qingwei XING, Man HUANG. The effect of different heteroatoms-doped Na2Ti3O7 on sodium ion storage [J]. Energy Storage Science and Technology, 2023, 12(9): 2715-2726. |
| [4] | Yinchen YANG, Shanling REN, Zhihong YANG, Yunhui WANG. First principles study of two-dimensional boron antimony films as anchoring materials for lithium-sulfur batteries [J]. Energy Storage Science and Technology, 2023, 12(9): 2760-2766. |
| [5] | Zhengguang ZHAO, Zhenying CHEN, Guangqun ZHAI, Xi ZHANG, Xiaodong ZHUANG. Preparation of Sc/O-doped sulfide electrolyte for all-solid-state batteries [J]. Energy Storage Science and Technology, 2023, 12(8): 2412-2423. |
| [6] | Zinan ZHANG, Jian CHEN. Preparation and property evaluation of Nb-doped Na3V2O2 (PO4 ) 2F hollow microspheres as cathode materials for sodium-ion batteries [J]. Energy Storage Science and Technology, 2023, 12(8): 2370-2381. |
| [7] | Lei LEI, Peng GAO, Nana FENG, Kunpeng CAI, Hai ZHANG, Yang ZHANG. The influences of multifactors in the synthesis progress on the characteristics of lithium lanthanum zirconate solid electrolytes [J]. Energy Storage Science and Technology, 2023, 12(5): 1625-1635. |
| [8] | Yongli YI, Ran YU, Wu LI, Yi JIN, Zheren DAI. Preparation of Mo, Al-doped Li7La3Zr2O12-based composite solid electrolyte and performance of all-solid-state batterys [J]. Energy Storage Science and Technology, 2023, 12(5): 1490-1499. |
| [9] | Wenzhe HAN, Qingsong LAI, Xuanwen GAO, Wenbin LUO. Advances toward manganese-based layered oxide cathodes for potassium-ion batteries [J]. Energy Storage Science and Technology, 2023, 12(5): 1364-1379. |
| [10] | Deliu ZHANG, Yan ZHANG, Hai WANG, Jiadong WANG, Xuanwen GAO, Chaomeng LIU, Dongrun YANG, Wenbin LUO. Optimization of high nickel cathode materials for lithium ion batteries by magnesium doped heterogeneous aluminum oxide coating [J]. Energy Storage Science and Technology, 2023, 12(2): 339-348. |
| [11] | Liyuan SHEN, Guixin ZHANG, Zhaoling MA. Catalytic conversion performance study of O-doped NiCo2S4/CNT composites for Li polysulfides [J]. Energy Storage Science and Technology, 2023, 12(11): 3318-3329. |
| [12] | Kejun CHEN, Lijun FAN. Controllable synthesis of Co2+-doped FeS2 and their sodium storage performances [J]. Energy Storage Science and Technology, 2023, 12(10): 3056-3063. |
| [13] | Liang WANG, Xin LIU, Changan WANG, Shengnian TIE. Preparation and thermal performance of nitrogen-doped porous carbon sponge-type mirabilite-based composite phase-change material [J]. Energy Storage Science and Technology, 2023, 12(1): 79-85. |
| [14] | Wenshu ZHANG, Fangyuan HU, Hao HUANG, Xudong WANG, Man YAO. Sodium storage anode based on titanium-based MXene and its performance regulation mechanism [J]. Energy Storage Science and Technology, 2023, 12(1): 35-41. |
| [15] | Kai ZHANG, Youlong XU. Research progress and development trend of sodium manganate cathode materials for sodium ion batteries [J]. Energy Storage Science and Technology, 2023, 12(1): 86-110. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
